b站生信课程02-1
r-bioinfolessonb站生信课程TCGA及GEO数据挖掘入门必看P3-P7笔记——数据下载与处理部分
写在前面:本篇教程来自b站课程TCGA及GEO数据挖掘入门必看 P3-P7 相关资料下载
数据下载
TCGA数据
进入TCGA官网
具体方法在b站生信课01中
Cohort Builder:
-
Program–TCGA -
Project–TCGA LUSC(肺鳞癌)
表达数据
Repository:
-
Data Category–transcriptome profiling -
Data Type–Gene Expression Quantification
Add All Files to Cart加数据添加到仓库,共553个
点击右上角的cart查看仓库,共下载两个文件,一个是cart
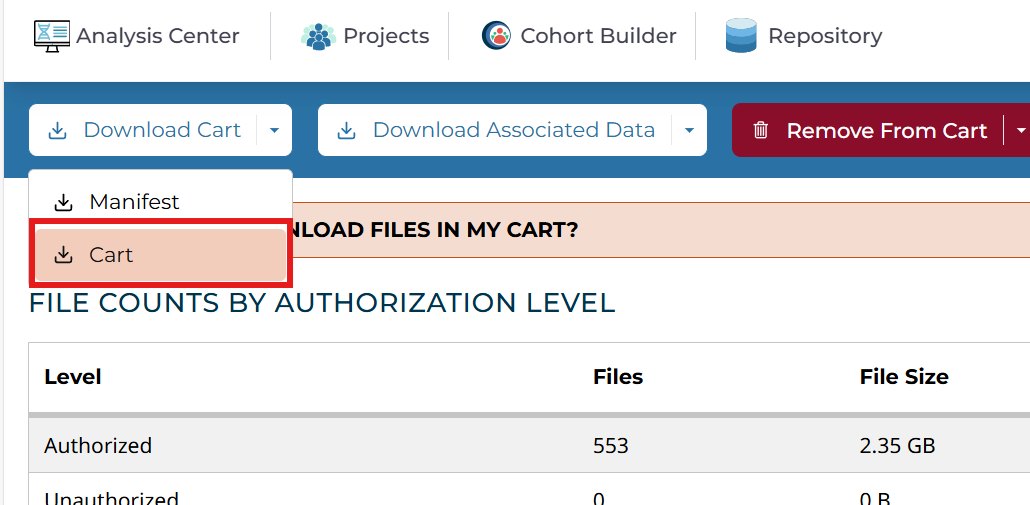
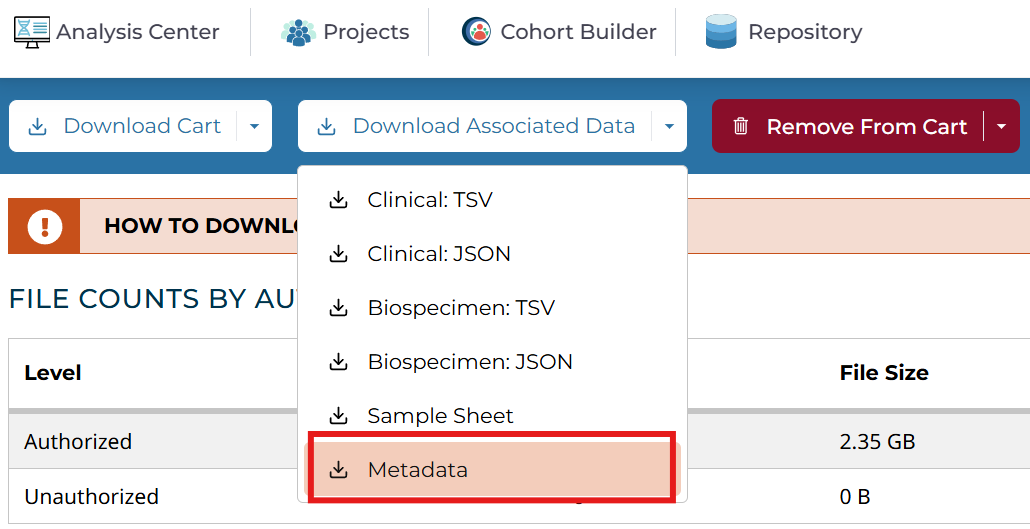
另一个是Metadata,它将文件名与样本名进行对应
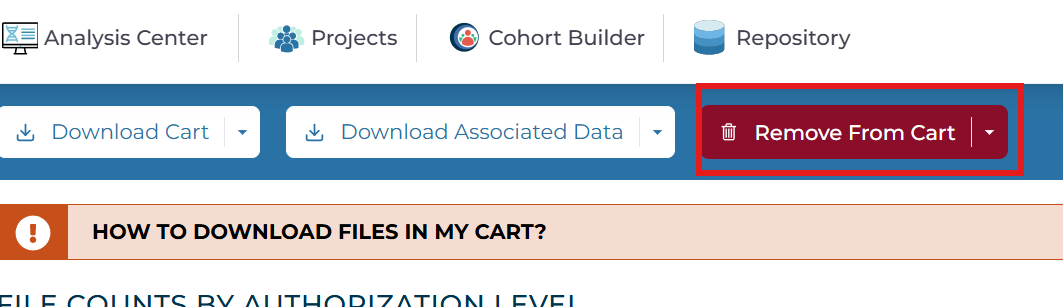
之后点击Remove From Cart将cart中文件清除,返回Repository,将其中的筛选项也重置,建议从下往上进行重置
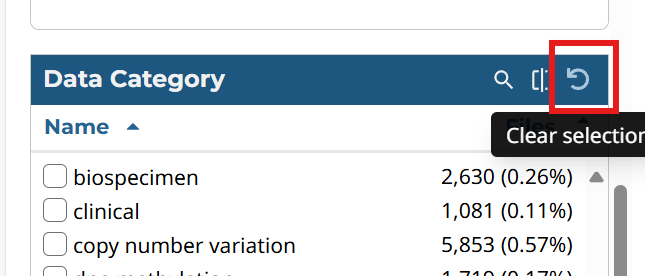
每次下载数据前都需清除原先的数据,下面不再重述
miRNA数据
Repository:
-
Data Category–transcriptome profiling -
Data Type–Isoform Expression Quantification
Add All Files to Cart加数据添加到仓库,共523个
下载cart和Metadata
临床数据
Repository:
-
Data Category–clinical -
Data Type–Clinical Supplement -
Data Format–bcr xml
Add All Files to Cart加数据添加到仓库,共504个
只需下载cart文件
其它数据
突变数据
Repository:
-
Data Category–simple nucleotide variation -
Data Type–Masked Somatic Mutation
Add All Files to Cart加数据添加到仓库,共549个
只需下载cart
甲基化数据
Repository:
-
Data Category–dna methylation -
Data Type–Methylation Beta Value -
Platform–illumina human methylation 450
Add All Files to Cart加数据添加到仓库,共412个
下载cart和Metadata
补充:癌症名称简写对照
另外的数据下载方式:
这两种都可以提供处理较完全的数据,但数据较旧
GEO数据
进入GEO官网
表达矩阵+注释文件
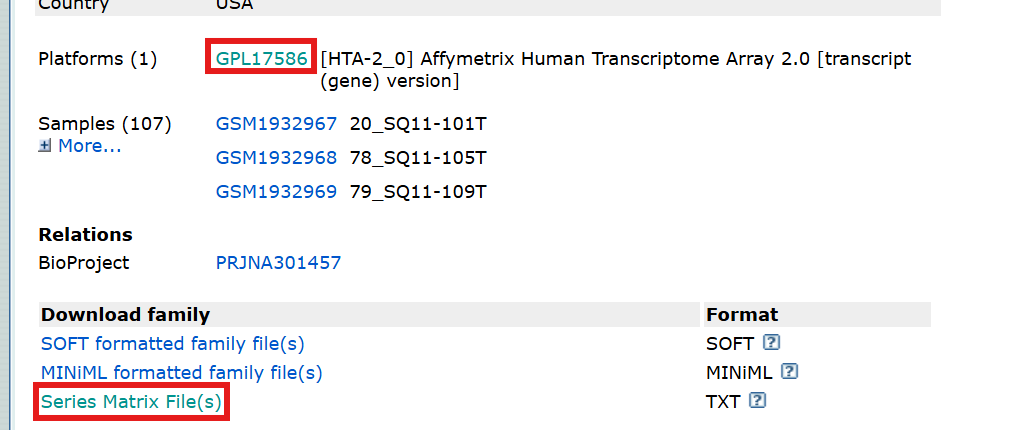
搜索数据集GSE74777,下载两个文件
一个是Series Matrix File(s)(表达矩阵),下载
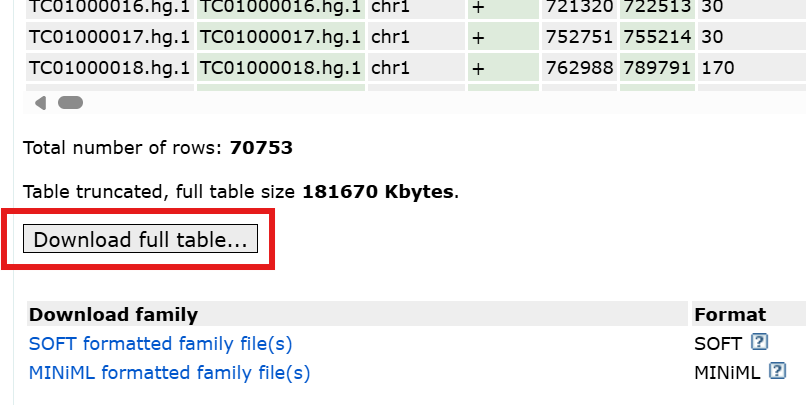
另一个是Platforms–GPL17586->Download full table...(注释文件)
表达矩阵+soft文件
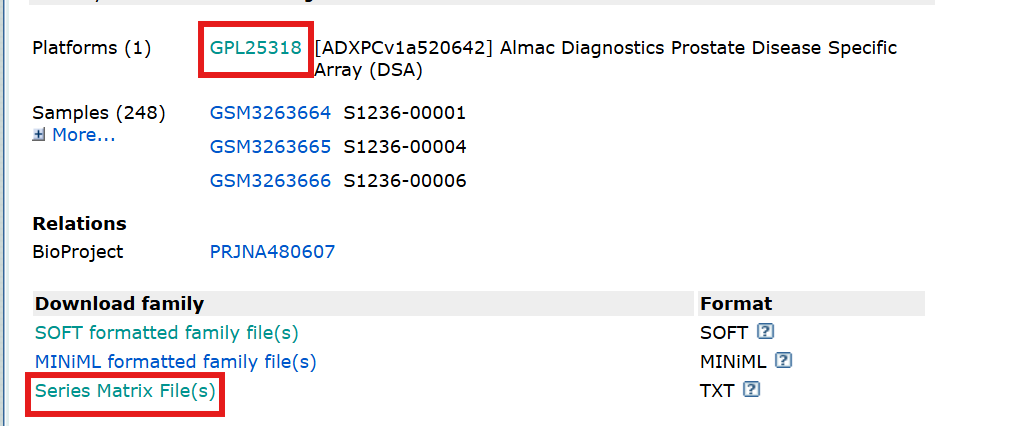
搜索数据集GSE116918,下载两个文件
一个是Series Matrix File(s)(表达矩阵),下载
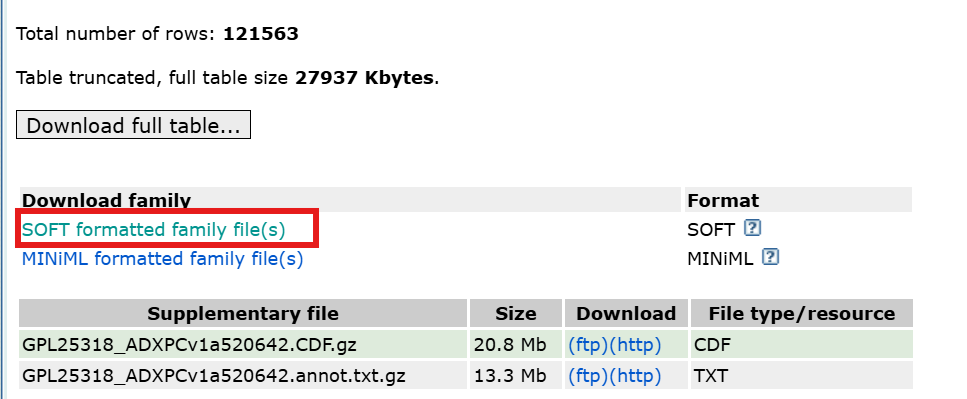
另一个是Platforms–GPL25318->SOFT formatted family file(s)(soft文件),下载
使用R包
搜索数据集GSE30219,只需下载Series Matrix File(s)(表达矩阵),下载
数据预处理
TCGA数据
表达数据
将下载的压缩包解压成文件夹,里面有553个文件夹和1个.txt文件(无用),每个文件夹内都有一个样本(tsv文件),格式如下
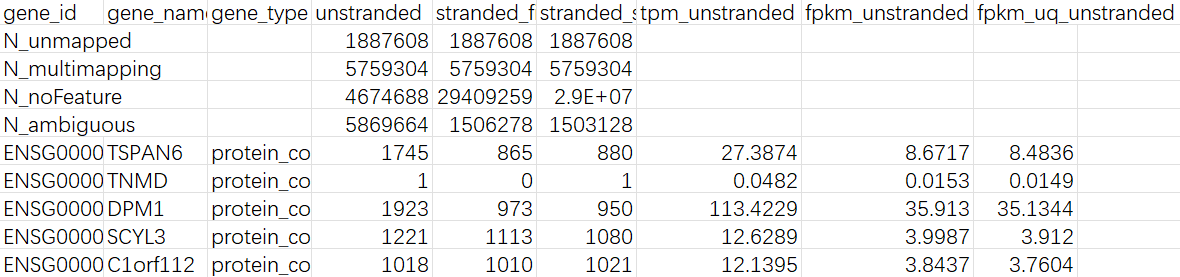
-
unstranded–count -
gene_type–基因类型 -
tpm_unstranded–TPM -
fpkm_unstranded–FPKM
可以发现缺少了该数据对应的样本名称,它存储在下载的metadata文件中
关键:将样本名称添加到表达数据中
读取metadata文件:使用rjson包,需要R版本>=4.4.0
if(!require("rjson", quietly = T))
{
install.packages("rjson");
library("rjson");
}
json <- jsonlite::fromJSON("data\\表达数据\\metadata.cart.2024-09-02.json");
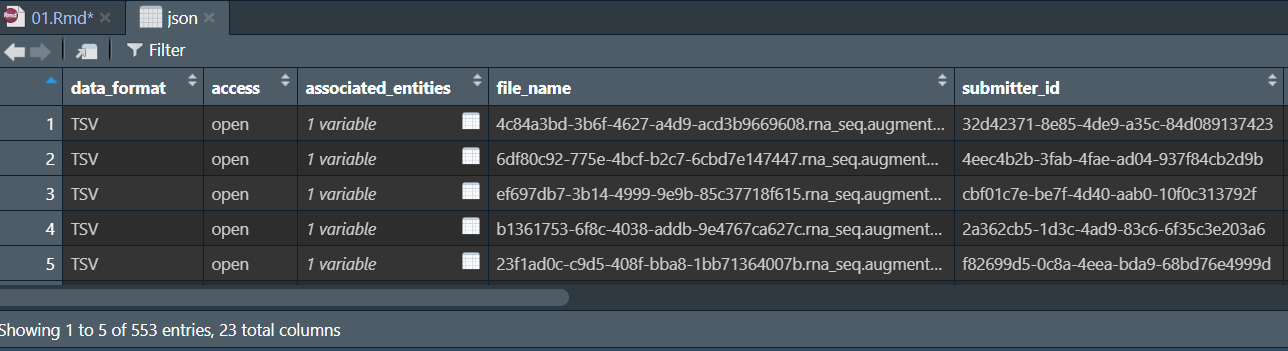
其中file_id是文件夹的名称,associated_entities这个表格中entity_submitter_id即为该文件夹中数据对应的样本名
# 样本名称,因为associated_entities的列值是一个list,不能直接用索引取,而是要遍历每个元素
sample_id <- sapply(json$associated_entities, function(x){x[,1]});
# 文件名称
file_name <- json$file_id;
# 将它们合并成一个df
file_sample <- data.frame(sample_id, file_name);
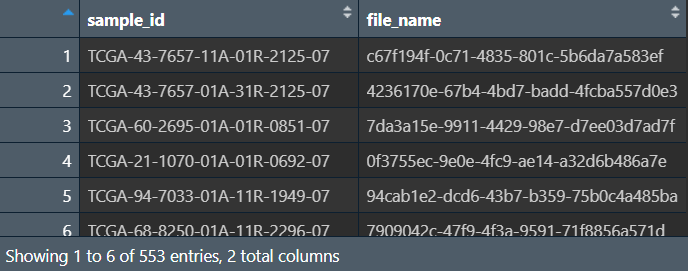
读取count数据:因为count数据都是在文件夹中,且数量很多,无法用常规方法读取
解决方法:使用list.files()函数,它可以遍历整个文件夹,读取指定后缀的文件,返回文件相对路径
# 数据所在文件夹
wd <- "data/表达数据/gdc_download_20240902_050017.880821/";
# 文件相对路径
count_file <- list.files(
wd,
pattern = '.tsv', # 指定读取tsv文件
recursive = T # 递归遍历(读取所有子文件)
);
# 文件绝对路径
count_file_path <- paste0(wd, count_file);
# 文件夹名:将路径按'/'切分,取第二个元素即为文件夹名
# 注意不是tsv文件的名称,而是它所在文件夹的名称
count_file_name <- strsplit(count_file, split = "/");
count_file_name <- sapply(count_file_name, function(x){x[1]});
# 注:此处function(x){x[1]}中取的是x[1],要根据自己的实际路径判断取x的第几个元素
根据上面得到的路径读取文件(使用循环)
-
因为我们仅使用tpm进行计算,所以只保留tpm列
-
根据文件夹名,确认其中数据的样本名(
file_sample中的sample_id),将其作为tpm列的列名 -
因为所有组数据的行名及其顺序都相同,直接列合并即可
# 结果矩阵
matrix <- data.frame(matrix(nrow = 60660, ncol = 0));
# 因为有60660个基因,所以有60660行
for (i in 1:length(count_file_path)) {
# 读取文件
count_data <- read.delim(count_file_path[i], fill = T, header = F, row.names = 1);
# 对读取结果进行一些更改
colnames(count_data) <- count_data[2, ]; # 更改列名为第二行的元素
count_data <- count_data[-c(1:6), ]; # 去除前6行无用数据(因为列名在第2行,所以不在读取时跳过)
count_data <- count_data[6]; # 取tpm列,如果取count列就是count_data[3]
# 该文件对应file_sample中的第几行样本
file_sample_index <- which(file_sample$file_name == count_file_name[i]);
# 将这组数据的列名改为样本名
colnames(count_data) <- file_sample$sample_id[file_sample_index];
# 将数据添加到结果矩阵中
matrix <- cbind(matrix, count_data);
}
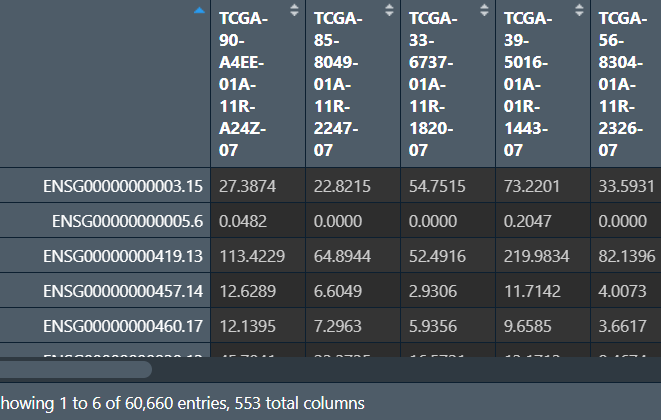
此时每组数据的行名ENSGxxx是基因名,但我们需要的是TNMD这种symbol形式的,需要进行转化
方法:因为所有tsv文件的这两列(两种基因名gene_name和gene_id)都相同,结果矩阵matrix的行名及其顺序也与每个tsv文件相同,所以随意取一个tsv文件的gene_name列,与matrix进行列合并即可。为方便后续进一步筛选,再将gene_type列也合并到结果中
为什么不直接将symbol作行名:在生信课01中提过,symbol有重复,无法作行名,后面需要去重处理后再作行名
count_data_single <- read.delim(count_file_path[1], fill = T, header = F, row.names = 1);
gene_name <- count_data_single[-c(1:6), 1];
gene_type <- count_data_single[-c(1:6), 2];
new_matrix <- cbind(gene_name, matrix);
new_matrix <- cbind(gene_type, new_matrix);
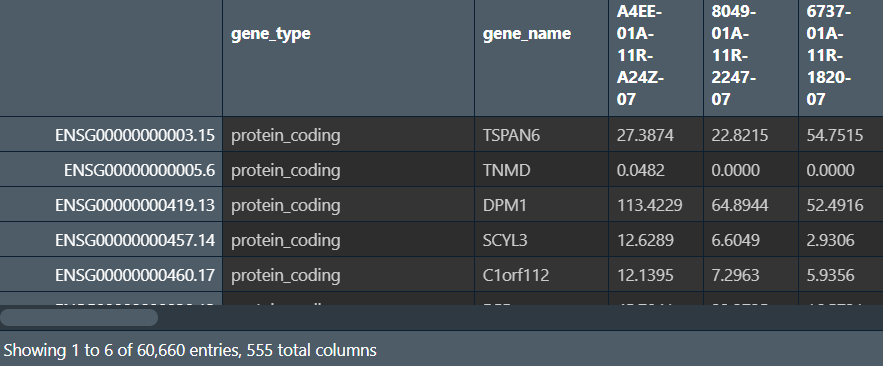
去除重复的基因:与生信课01中方法相同,都是使用aggregate函数,这里保留tpm最大的基因
new_matrix <- aggregate(.~gene_name, data = new_matrix, FUN = max);
只保留mRNA基因:根据gene_type==”protein_coding”筛选
new_matrix <- subset(x = new_matrix, gene_type=="protein_coding");
将gene_name列作为行名:
rownames(new_matrix) <- new_matrix[, 1];
new_matrix <- new_matrix[, -c(1,2)]; # 删除这两列
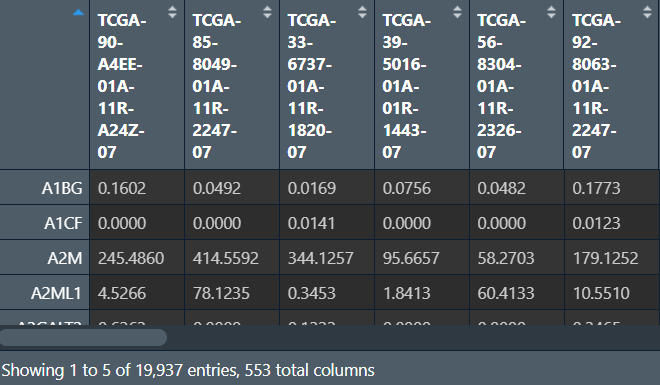
保存到文件中:
-
将行名(
gene_name)单独作一列 -
将列名中的
.改为-(data.frame函数会自动将列名中的-改为.,需要再改回去)
save_matrix <- data.frame(
ID=rownames(new_matrix),
new_matrix
);
# 注意:正则表达式转义,'.'要写成'[.]'
colnames(save_matrix) <- gsub('[.]', '-', colnames(save_matrix));
# 保存数据
write.table(
save_matrix,
"save_data\\TCGA_LUSC_TPM.txt",
sep = '\t',
quote = F,
row.names = F
);
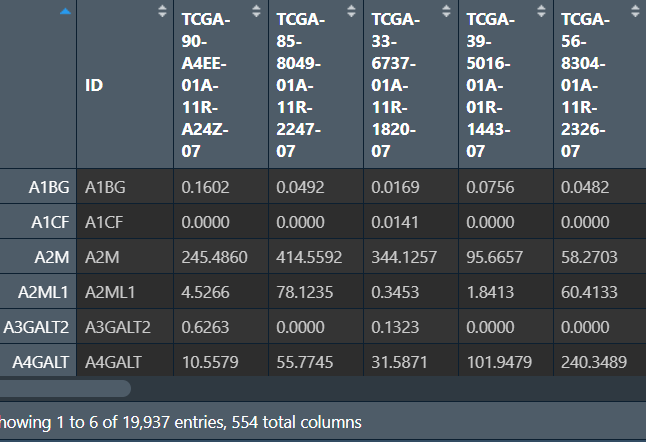
代码汇总:
rm(list=ls());
if(!require("rjson", quietly = T))
{
install.packages("rjson");
library("rjson");
}
json <- jsonlite::fromJSON("data\\表达数据\\metadata.cart.2024-09-02.json");
sample_id <- sapply(json$associated_entities, function(x){x[,1]});
file_name <- json$file_id;
file_sample <- data.frame(sample_id, file_name);
wd <- "data/表达数据/gdc_download_20240902_050017.880821/";
count_file <- list.files(wd, pattern = '.tsv', recursive = T);
count_file_path <- paste0(wd, count_file);
count_file_name <- strsplit(count_file, split = "/");
count_file_name <- sapply(count_file_name, function(x){x[1]});
matrix <- data.frame(matrix(nrow = 60660, ncol = 0));
for (i in 1:length(count_file_path)) {
count_data <- read.delim(count_file_path[i], fill = T, header = F, row.names = 1);
colnames(count_data) <- count_data[2, ];
count_data <- count_data[-c(1:6), ];
count_data <- count_data[6];
file_sample_index <- which(file_sample$file_name == count_file_name[i]);
colnames(count_data) <- file_sample$sample_id[file_sample_index];
matrix <- cbind(matrix, count_data);
}
count_data_single <- read.delim(count_file_path[1], fill = T, header = F, row.names = 1);
gene_name <- count_data_single[-c(1:6), 1];
gene_type <- count_data_single[-c(1:6), 2];
new_matrix <- cbind(gene_name, matrix);
new_matrix <- cbind(gene_type, new_matrix);
new_matrix <- aggregate(.~gene_name, data = new_matrix, FUN = max);
new_matrix <- subset(x = new_matrix, gene_type=="protein_coding");
rownames(new_matrix) <- new_matrix[, 1];
new_matrix <- new_matrix[, -c(1,2)];
save_matrix <- data.frame(ID=rownames(new_matrix), new_matrix
);
colnames(save_matrix) <- gsub('[.]', '-', colnames(save_matrix));
write.table(save_matrix, "save_data\\TCGA_LUSC_TPM.txt", sep = '\t', quote = F, row.names = F);
临床数据
读取xml格式数据:使用XML包
if(!require("XML", quietly = T))
{
install.packages("XML");
library("XML");
}
# 与上节课相同方法先获取文件路径
wd <- "data/临床数据/gdc_download_20240902_053601.073446"
file_names <- list.files(
path = wd,
pattern = '.xml', # 后缀为xml的文件
recursive = T
);
这里使用一种与上节课不同的方法遍历file_names读取文件
-
使用
lapply函数,遍历file_names,将读取的数据(一个df)保存到一个列表中 -
使用
do.call函数,将列表中的df合并
具体的读取方式:
-
完整路径拼接:使用
file.path(绝对路径, 相对路径),它会自动补齐两个路间的/ -
xmlParse函数读取xml文件 -
xmlRoot函数获取xml文件的所有内容 -
xmlToDataFrame函数将指定xml内容转为df -
返回转置后结果
df_list <- lapply(file_names, function(x){
file_path <- file.path(wd, x); # 完整路径
res <- xmlParse(file = file_path); # xml文件
rootnode <- xmlRoot(res); # xml文件内容
xml_df <- xmlToDataFrame(rootnode[2]); # 转为df
return(xml_df); # 返回结果
});
data <- do.call(rbind, df_list); # 合并
每个df格式:
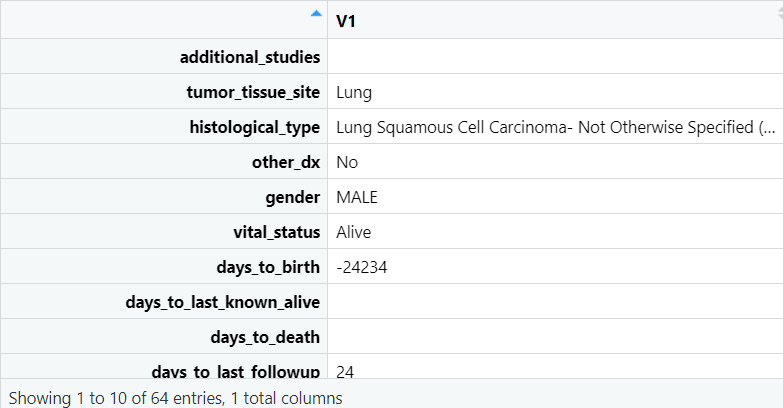
最终结果:
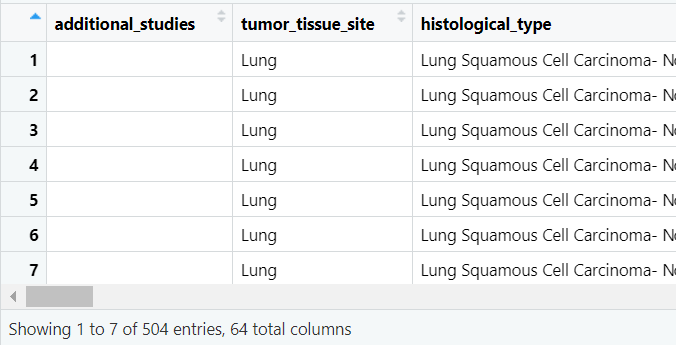
数据处理:
-
删除前3列(病变部位、病名称等),这些信息我们已知
-
计算生存时间
-
如果还存活,就以最后一次随访的时间
days_to_last_followup为生存时间 -
如果已死亡,就以死亡时间
days_to_death为生存时间 -
最后删除
days_to_last_known_alive和上面那两列
-
-
修改临床分期
stage_event的列值,列值的含义:”Stage IVT4N1M1”中”Stage IV”就是肿瘤分期,T/N/M是更具体的分期-
删除”3rd”/”4th”/…
-
如果不是以
Stage开头,就在前面加上Stage X
-
new_data <- data[, -c(1:3)]; # 删除前3列
new_data$survival_time <- ifelse(
new_data$vital_status=='Alive',
new_data$days_to_last_followup, # 如果存活,以days_to_death为列值
new_data$days_to_death # 如果死亡,以days_to_death为列值
);
new_data <- new_data[, c(ncol(new_data), 1:(ncol(new_data)-1))]; # 将最后一列survival_time移到第一列
del_col <- c("days_to_last_followup", "days_to_death", "days_to_last_known_alive"); # 要删除的列
library("dplyr");
new_data <- dplyr::select(new_data, -del_col); # 删除这3列
del_strs <- c("3rd", "4th", "5th", "6th", "7th"); # 要删除的字符
for(del_str in del_strs){ # 进行替换(删除)
new_data$stage_event <- gsub(del_str, "", new_data$stage_event);
}
new_data$stage_event <- ifelse(
startsWith(new_data$stage_event, 'Stage'),
new_data$stage_event, # 如果以"Stage"开头就不变
paste0("Stage X", new_data$stage_event) # 反之加上"Stage X"
);
# 保存数据,使用writexl包保存成xlsx格式
if(!require("writexl", quietly = T))
{
install.packages("writexl");
library("writexl");
}
write_xlsx(
new_data,
'save_data\\clinical.xlsx'
);
使用excel再次处理数据:将stage_event列按T/N/M分隔,分成4列
-
用excel打开刚才的xlsx文件,单击
stage_event列后面的那列(我这里是karnofsky_performance_score列)的列名AB,选中它,右键,插入,共插入3列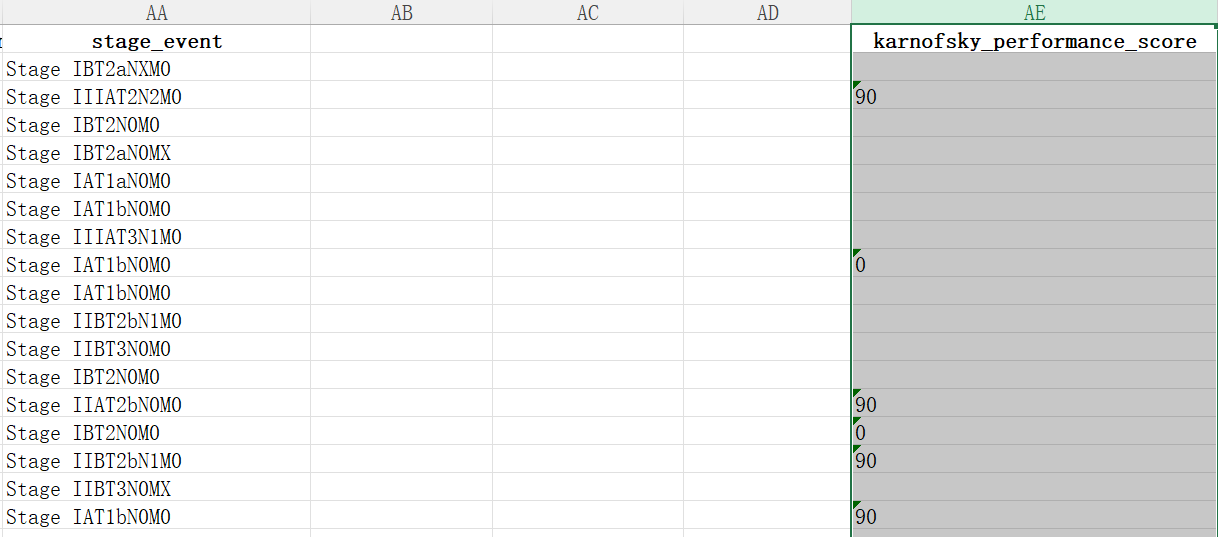
-
选中
stage_event列,点击数据中的分列,选中分隔符号,下一步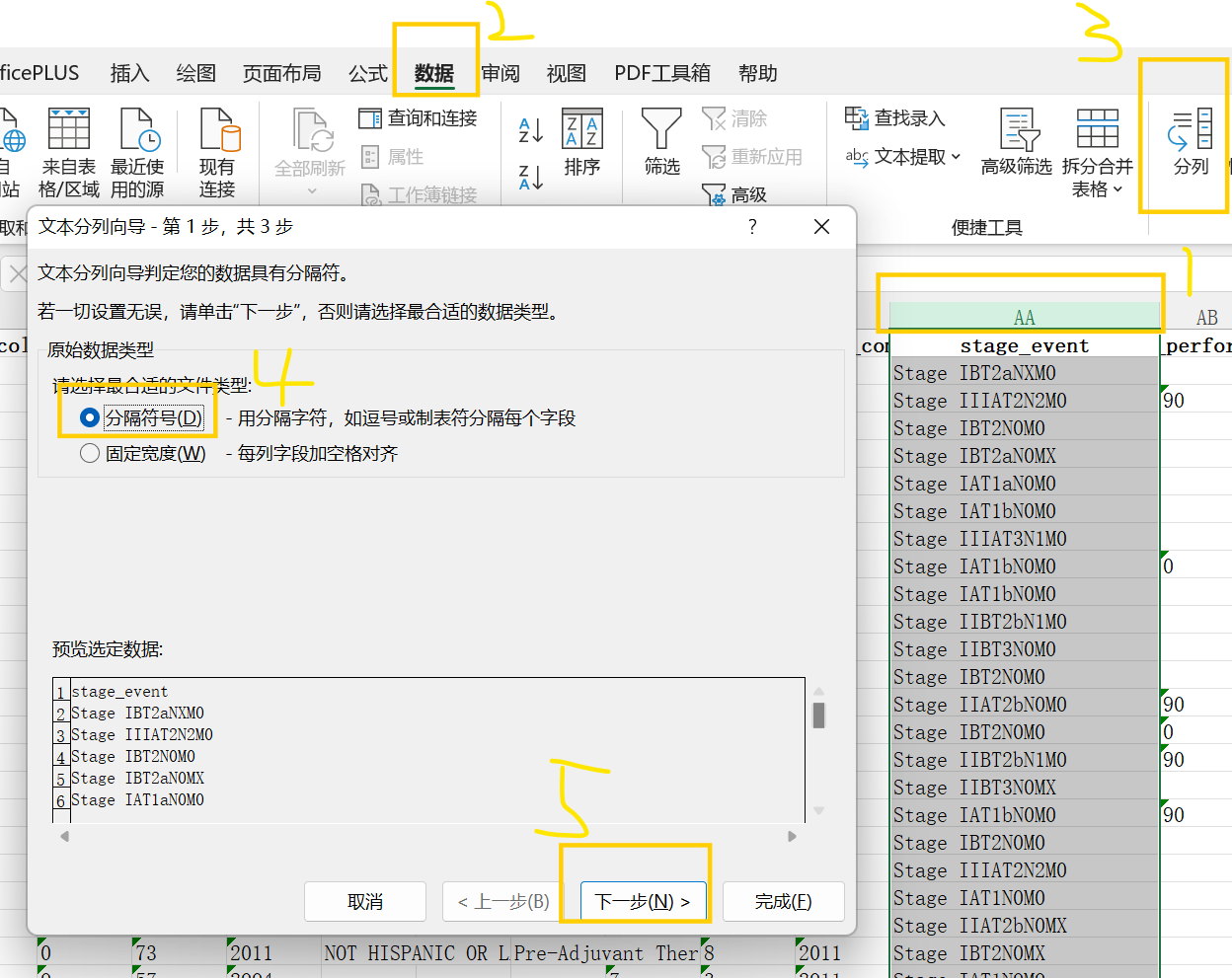
-
选定分隔符号为
其他,输入T,点击完成,即可按T进行分隔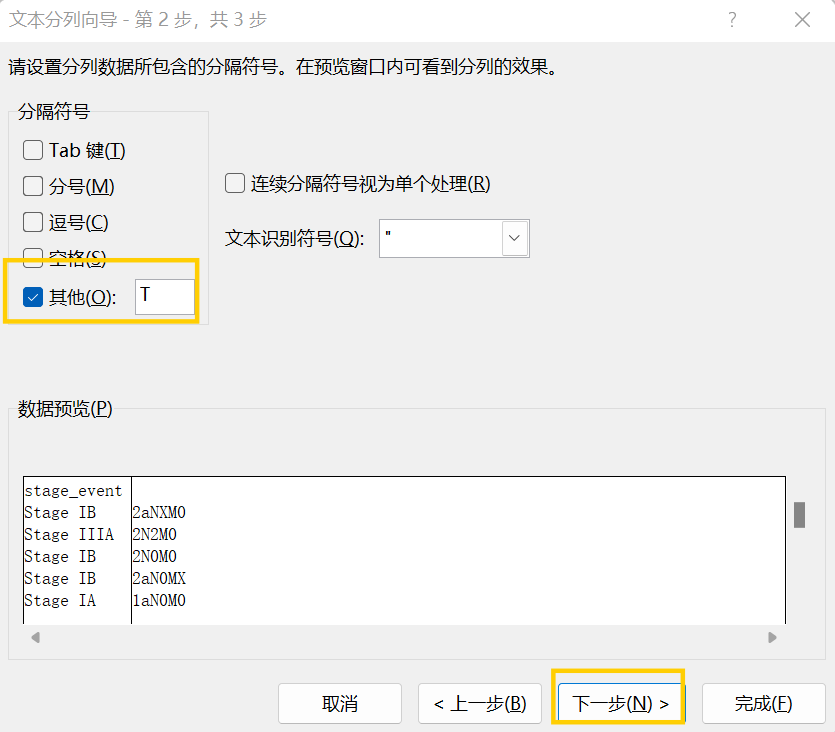
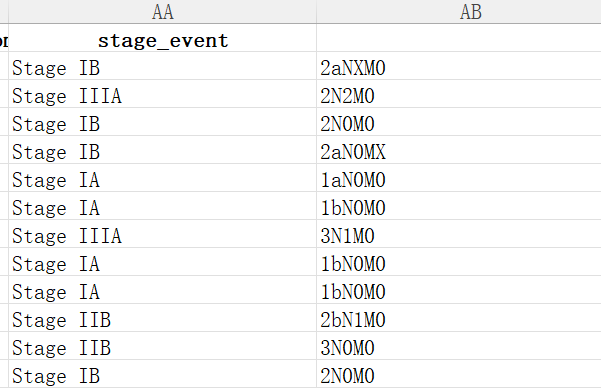
-
再用相同方法,选中刚刚分隔的列,依次按
N和M分隔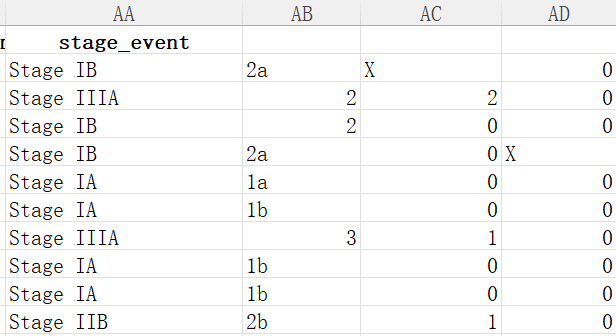
-
更改它们的列名分别为T/N/M

代码汇总:
rm(list=ls());
if(!require("XML", quietly = T))
{
install.packages("XML");
library("XML");
}
wd <- "data/临床数据/gdc_download_20240902_053601.073446"
file_names <- list.files(path = wd, pattern = '.xml', recursive = T);
df_list <- lapply(file_names, function(x){
file_path <- file.path(wd, x);
res <- xmlParse(file = file_path);
rootnode <- xmlRoot(res);
xml_df <- xmlToDataFrame(rootnode[2]);
return(xml_df);
});
data <- do.call(rbind, df_list);
new_data <- data[, -c(1:3)];
new_data$survival_time <- ifelse(
new_data$vital_status=='Alive',
new_data$days_to_last_followup,
new_data$days_to_death
);
new_data <- new_data[, c(ncol(new_data), 1:(ncol(new_data)-1))];
del_col <- c("days_to_last_followup", "days_to_death", "days_to_last_known_alive");
library("dplyr");
new_data <- dplyr::select(new_data, -del_col);
del_strs <- c("3rd", "4th", "5th", "6th", "7th");
for(del_str in del_strs){
new_data$stage_event <- gsub(del_str, "", new_data$stage_event);
}
new_data$stage_event <- ifelse(
startsWith(new_data$stage_event, 'Stage'),
new_data$stage_event,
paste0("Stage X", new_data$stage_event)
);
if(!require("writexl", quietly = T))
{
install.packages("writexl");
library("writexl");
}
write_xlsx(new_data, 'save_data\\clinical.xlsx');
miRNA数据
需要包:tidyverse、BiocManager、miRBaseVersions.db
if(!require("tidyverse", quietly = T))
{
install.packages("tidyverse");
library("tidyverse");
}
if(!require("BiocManager", quietly = T))
{
install.packages("BiocManager");
library("BiocManager");
}
if(!require("miRBaseVersions.db", quietly = T))
{
BiocManager::install("miRBaseVersions.db");
library("miRBaseVersions.db");
}
# 获取所有的文件名
wd <- "data/miRNA数据/gdc_download_20240902_053252.211289/"
file_names <- list.files(
wd,
pattern = 'isoforms.quantification.txt',
recursive = T
);
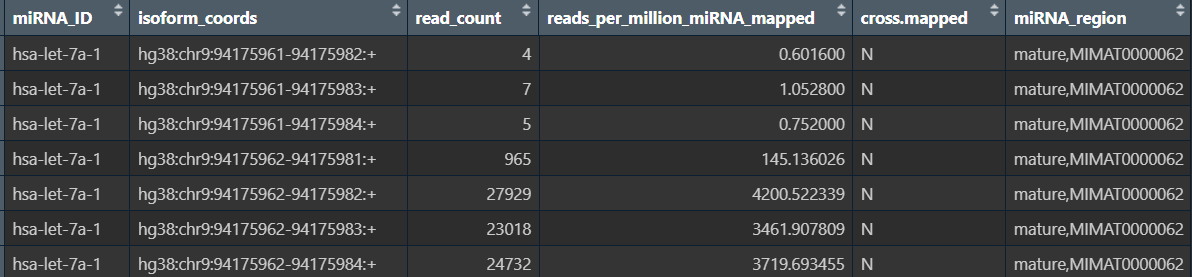
数据读取与初处理:
-
只取第4列(RPKM值)和第6列(miRNA名称)
-
将相同的
miRNA_region对应的RPKM值加和:按miRNA_region分组,之后summarise
df_list <- list(); # 初始化结果列表
for (i in 1:length(file_names)) {
df <- read.table(
file.path(wd, file_names[i]),
sep = '\t',
header = T
);
df_list[[i]] <- df %>% # 添加到结果中
dplyr::select(c(6, 4)) %>% # 只取第4、6列
group_by(miRNA_region) %>% # 按区域分组
summarise(reads_per_million_miRNA_mapped = sum(reads_per_million_miRNA_mapped)); # 加和
}
每个df:
更改后的每个df:
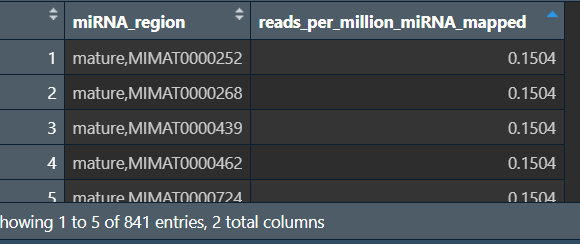
想实现的效果:
-
miRNA_region作行名 -
每一列都是一个样本的RPKM值
reads_per_million_miRNA_mapped
注意:原数据中,RPKM值为0的行被删除了,这导致每个df的行数不同,不能直接进行用cbind合并
方法:使用merge函数,将两个df按miRNA_region列进行合并,该函数会自动将空缺值补成NA;配合Reduce函数,遍历df_list
data <- Reduce(function(x, y){
merge(x, y, by = 'miRNA_region', all = T);
}, df_list); # 合并
data[is.na(data)] <- 0; # NA->0
data <- column_to_rownames(data, var = 'miRNA_region'); # 列变行名
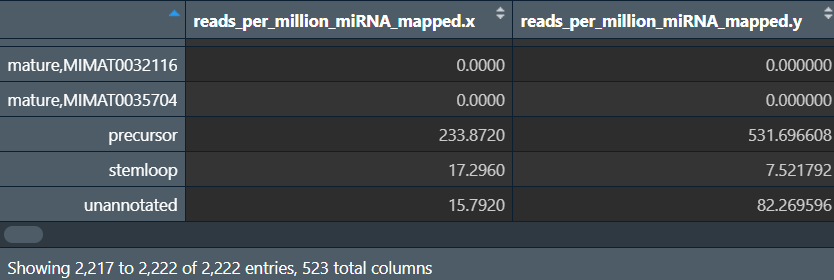
其它处理:
-
删去最后三行无用数据
-
删去行名中的
mature,
data <- data[-((nrow(data)-2):nrow(data)), ]; # 删去最后三行
rownames(data) <- str_remove(rownames(data), "mature,"); # 更改行名
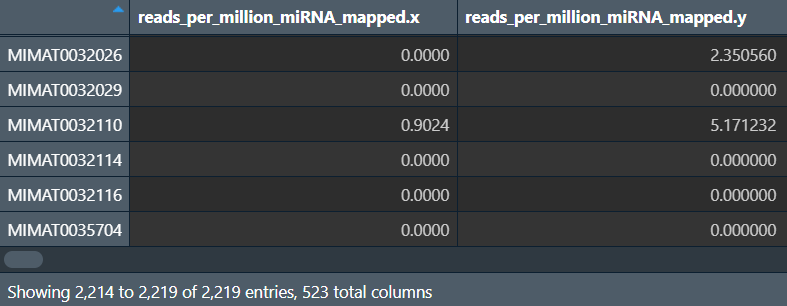
miRNA名称(行名)修改:现在的基因名称(行名)都是以MIM开头(ACCESSION类型的基因id),需要转换为以hsa-miR开头的成熟体id(NAME类型的基因id)
mh <- select(
miRBaseVersions.db, # 使用的数据库
keys = rownames(data), # 数据的基因名称
keytype = "MIMAT", # 数据的基因类型
columns = c("ACCESSION", "NAME", "VERSION") # 要转换的基因类型、数据的基因类型、版本号
);
# 因为原数据使用的gdc数据库是mirBase21版本,所以只选出版本号为21的行
mh <- mh[mh$VERSION=='21', ];
# 对mh调整顺序,使其行顺序与data相同
mh <- mh[match(rownames(data), mh$ACCESSION), ]; # match(x, y)函数返回x在y中的索引
rownames(data) <- mh$NAME; # 更改行名
注:这里完成的任务类似于生信课01中org.Hs.eg.db的mapIds函数转换
mh:
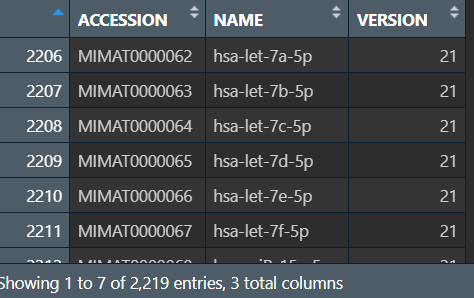
data:
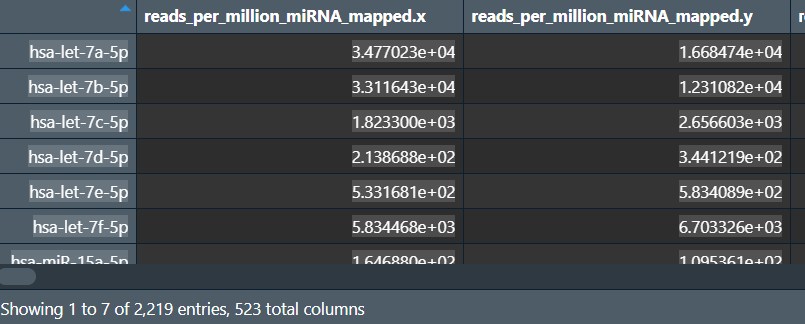
行名中-3p/-5p是3’/5’端的意思
样本名(列名)修改:
使用读入metadata文件,方法与表达数据中的相同
library("rjson");
json <- jsonlite::fromJSON("data\\miRNA数据\\metadata.cart.2024-09-02.json");
sample_id <- sapply(json$associated_entities, function(x){x$entity_submitter_id}); # 样本名称
json_file_name <- json$file_id; # 文件名称
file_sample <- data.frame(sample_id, json_file_name); # 将它们合并成一个df
data_file_names <- strsplit(file_names, split = "/");
data_file_names <- sapply(data_file_names, function(x){x[1]}); # data中的文件名称
# 对file_sample调整顺序,使其行顺序与data_file_names相同
file_sample <- file_sample[match(data_file_names, file_sample$json_file_name), ];
colnames(data) <- file_sample$sample_id; # 更改列名
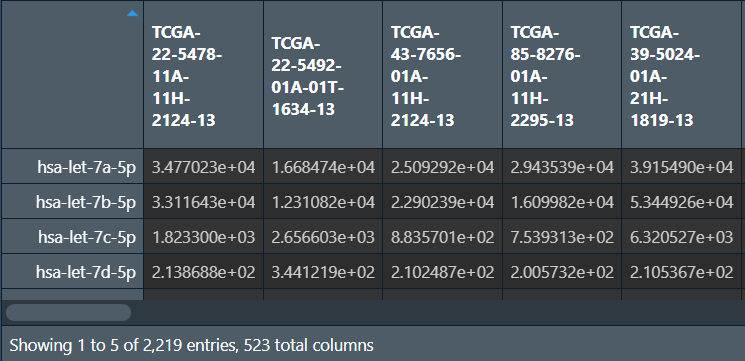
过滤:对于一个基因,如果有100个以上样本对它的表达量>0,就保留
方法:使用apply函数,设置其MARGIN参数为1(遍历行),自定义一个处理函数function(x),如果sum(x>0)>100就返回T。该函数最终返回一个bool值数组,表示某行是否满足要求,之后根据bool值取行即可
原理:x>0表示x值是否>0,如果>0就返回T,sum(x>0)中,T被转换成数值1,如果该行有100个值>0,就有100个T相加,结果为100
index <- apply(data, 1, function(x){
sum(x>0) > 100;
}); # 某行是否满足条件
new_data <- data[index, ]; # 根据bool值取行
# new_data <- as.matrix(new_data);
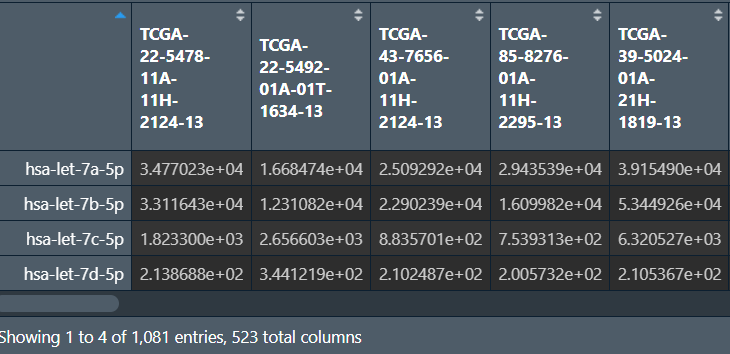
保存数据:
-
将行名作为一列
-
将列名中的
.替换为-
save_data <- data.frame(
ID = rownames(new_data), # 将行名作为一列
new_data
);
colnames(save_data) <- gsub('[.]', '-', colnames(save_data)); # 更改列名
write.table(
save_data,
"save_data\\miRNA.RPM.txt",
sep = '\t',
quote = F,
row.names = F
);
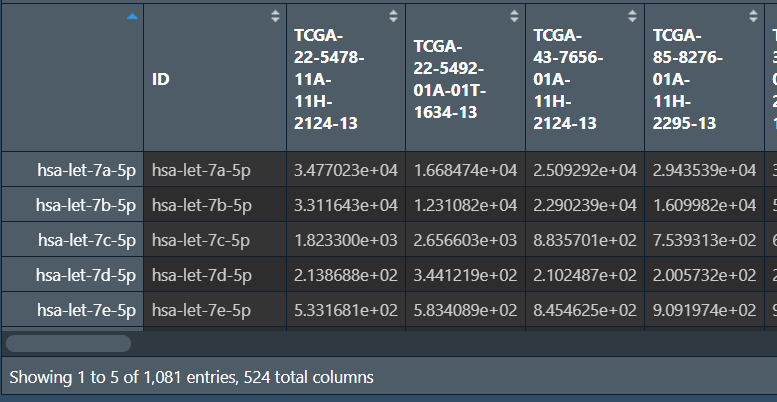
代码汇总:
rm(list=ls());
if(!require("tidyverse", quietly = T))
{
install.packages("tidyverse");
library("tidyverse");
}
if(!require("BiocManager", quietly = T))
{
install.packages("BiocManager");
library("BiocManager");
}
if(!require("miRBaseVersions.db", quietly = T))
{
library("BiocManager");
BiocManager::install("miRBaseVersions.db");
library("miRBaseVersions.db");
}
wd <- "data/miRNA数据/gdc_download_20240902_053252.211289/"
file_names <- list.files(wd, pattern = 'isoforms.quantification.txt', recursive = T);
df_list <- list();
for (i in 1:length(file_names)) {
df <- read.table(file.path(wd, file_names[i]), sep = '\t', header = T);
df_list[[i]] <- df %>%
dplyr::select(c(6, 4)) %>%
group_by(miRNA_region) %>%
summarise(reads_per_million_miRNA_mapped = sum(reads_per_million_miRNA_mapped));
}
data <- Reduce(function(x, y){merge(x, y, by = 'miRNA_region', all = T);}, df_list);
data[is.na(data)] <- 0;
data <- column_to_rownames(data, var = 'miRNA_region');
data <- data[-((nrow(data)-2):nrow(data)), ];
rownames(data) <- str_remove(rownames(data), "mature,");
mh <- select(
miRBaseVersions.db,
keys = rownames(data),
keytype = "MIMAT",
columns = c("ACCESSION", "NAME", "VERSION")
);
mh <- mh[mh$VERSION=='21', ];
mh <- mh[match(rownames(data), mh$ACCESSION), ];
rownames(data) <- mh$NAME;
library("rjson");
json <- jsonlite::fromJSON("data\\miRNA数据\\metadata.cart.2024-09-02.json");
sample_id <- sapply(json$associated_entities, function(x){x$entity_submitter_id});
json_file_name <- json$file_id;
file_sample <- data.frame(sample_id, json_file_name);
data_file_names <- strsplit(file_names, split = "/");
data_file_names <- sapply(data_file_names, function(x){x[1]});
file_sample <- file_sample[match(data_file_names, file_sample$json_file_name), ];
colnames(data) <- file_sample$sample_id;
index <- apply(data, 1, function(x){sum(x>0) > 100;});
new_data <- data[index, ];
save_data <- data.frame(ID = rownames(new_data), new_data);
colnames(save_data) <- gsub('[.]', '-', colnames(save_data));
write.table(save_data, "save_data\\miRNA.RPM.txt", sep = '\t', quote = F, row.names = F);
GEO数据
基础概念——探针:在基因表达检测中,用于检测基因的表达量。一般情况下,一个探针的id对应一种基因。在表达矩阵中,如果行名是探针id,就需要转为基因id以进一步分析
表达矩阵+注释文件
需要包:tidyverse、R.utils、GEOquery、
if(!require("GEOquery", quietly = T))
{
BiocManager::install("limma");
BiocManager::install("GEOquery");
library("GEOquery");
}
if(!require("R.utils", quietly = T))
{
install.packages('R.utils');
library("R.utils");
}
library("tidyverse");
读取数据–表达矩阵:与生信课01中的相同。使用getGEO函数,该函数既可以下载数据集,也可以读取数据集
注意:下载的.gz文件无需解压,下同
wd <- "data/GSE74777";
gset <- getGEO(
"GSE74777", # 数据集名称
destdir = wd, # 读取/下载路径
AnnotGPL = F,
getGPL = F # 读取/下载基因注释
);
dat <- exprs(gset[[1]]); # 获取表达矩阵
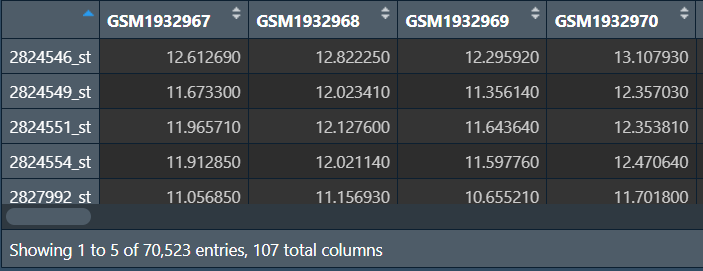
其中列名是样本名称,行名是探针id
可以看到表达矩阵的数据值不是count值(count是整数),要判断是否需要取log
以下函数实现这一功能:如果需要取log,它就会自动完成取log,并输出”finished”;反之输出”not needed”
get_log <- function(dat){
ex <- dat;
qx <- as.numeric(quantile(ex, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T));
LogC <- (qx[5]>100) ||
(qx[6]-qx[1]>50 && qx[2]>0) ||
(qx[2]>0 && qx[2]<1 && qx[4]>1 && qx[4]<2);
if(LogC){
ex[which(ex<=0)] <- NA;
ex <- log2(ex);
print("log2 transform finished");
}
else{
print("log2 transform not needed");
}
return(ex);
};
dat <- get_log(dat);
读取数据–临床信息:
pd <- pData(gset[[1]]);
# 导出
write.csv(pd, "data\\GSE74777\\clinical_GSE74777.csv");
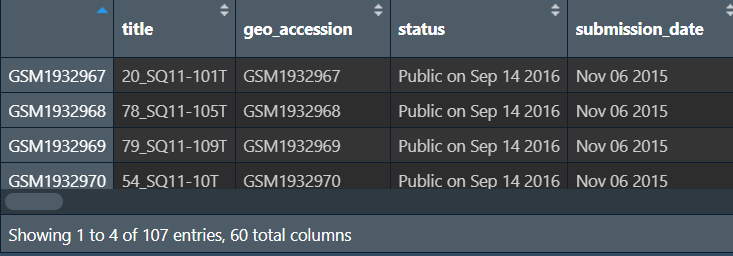
行名是样本名称,列名包括肿瘤信息、os、os_event等等
读取数据–注释文件:
gpl <- read.table(
"data\\GSE74777\\GPL17586-45144.txt",
header = T,
fill = T,
sep = '\t',
comment.char = '#',
stringsAsFactors = F,
quote = ""
);
ids <- gpl[, c("ID", "gene_assignment")]; # 只保留探针id和基因symbol列
colnames(ids) <- c('probe_id', 'symbol'); # 更改列名
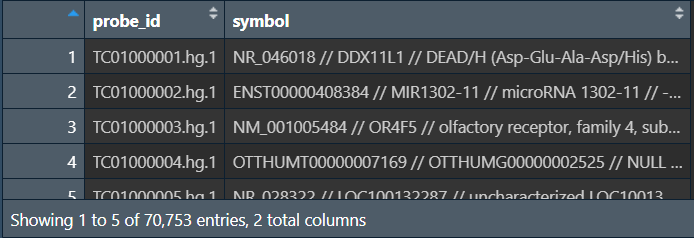
我们需要的基因symbol是symbol列按//分隔后的第二个元素,对于没有symbol的基因直接去掉
ids$symbol <- trimws( # 去掉字符串两端空格
str_split(ids$symbol, "//", simplify = T)[, 2] # 取第二个元素
);
ids <- ids[ids$symbol != '', ];
ids <- ids[ids$symbol != '---', ]; # 去掉没有symbol的基因
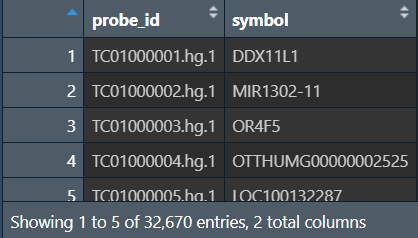
表达数据处理:
注释文件的作用:它的probe_id列与表达矩阵dat的行名对应(都是探针id),需要将dat的行名改为对应的基因symbol
ids <- ids[ids$probe_id %in% rownames(dat), ]; # 取出ids在dat行名中的探针id
dat <- dat[ids$probe_id, ]; # 根据行名(探针id)取出dat在ids探针id中的行
# 相当于根据探针id对ids和dat取交集,并让dat的行顺序与ids相同
table(rownames(dat)==ids$probe_id); # 查看探针id是否一致(全为T即为一致)
进行合并,同时去掉重复的基因,保留最大值
new_dat <- cbind(ids, dat);
new_dat <- aggregate(.~symbol, data=new_dat, FUN=max);
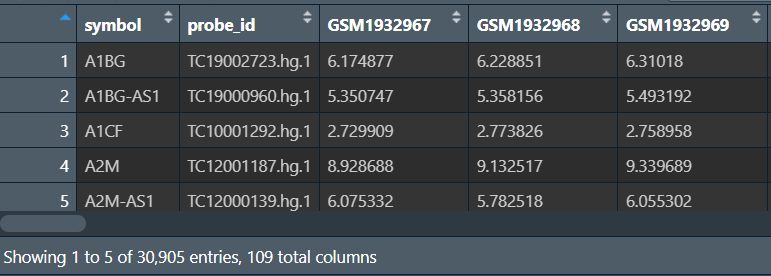
注:此处还要再查看一下是否还有symbol为空的行(看开头结尾就行)
去重后转化行名:
rownames(new_dat) <- new_dat[, 1]; # 改行名
new_dat <- new_dat[, -c(1, 2)]; # 去除前两列(探针id和基因名)
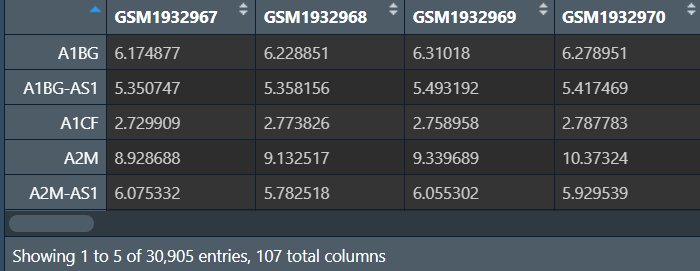
导出:
write.table(
data.frame(
ID=rownames(new_dat),
new_dat
),
file = "data\\GSE74777\\GSE74777.txt",
sep = '\t',
quote = F,
row.names = F
);
代码汇总:
rm(list=ls());
if(!require("GEOquery", quietly = T))
{
BiocManager::install("limma");
BiocManager::install("GEOquery");
library("GEOquery");
}
if(!require("R.utils", quietly = T))
{
install.packages('R.utils');
library("R.utils");
}
library("tidyverse");
wd <- "data/GSE74777";
gset <- getGEO("GSE74777", destdir = wd, AnnotGPL = F, getGPL = F);
dat <- exprs(gset[[1]]);
get_log <- function(dat){
ex <- dat;
qx <- as.numeric(quantile(ex, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T));
LogC <- (qx[5]>100) || (qx[6]-qx[1]>50 && qx[2]>0) || (qx[2]>0 && qx[2]<1 && qx[4]>1 && qx[4]<2);
if(LogC){
ex[which(ex<=0)] <- NA;
ex <- log2(ex);
print("log2 transform finished");
}
else{
print("log2 transform not needed");
}
return(ex);
};
dat <- get_log(dat);
pd <- pData(gset[[1]]);
write.csv(pd, "data\\GSE74777\\clinical_GSE74777.csv");
gpl <- read.table("data\\GSE74777\\GPL17586-45144.txt", header = T, fill = T, sep = '\t', comment.char = '#', stringsAsFactors = F, quote = "");
ids <- gpl[, c("ID", "gene_assignment")];
colnames(ids) <- c('probe_id', 'symbol');
ids$symbol <- trimws(str_split(ids$symbol, "//", simplify = T)[, 2]);
ids <- ids[ids$symbol != '', ];
ids <- ids[ids$symbol != '---', ];
ids <- ids[ids$probe_id %in% rownames(dat), ];
dat <- dat[ids$probe_id, ];
new_dat <- cbind(ids, dat);
new_dat <- aggregate(.~symbol, data=new_dat, FUN=max);
rownames(new_dat) <- new_dat[, 1];
new_dat <- new_dat[, -c(1, 2)];
write.table(data.frame(ID=rownames(new_dat),new_dat),
file = "data\\GSE74777\\GSE74777.txt", sep = '\t', quote = F, row.names = F);
表达矩阵+注释文件
与第一种方式相同的地方不再说明
# 读取表达矩阵和临床信息部分同上
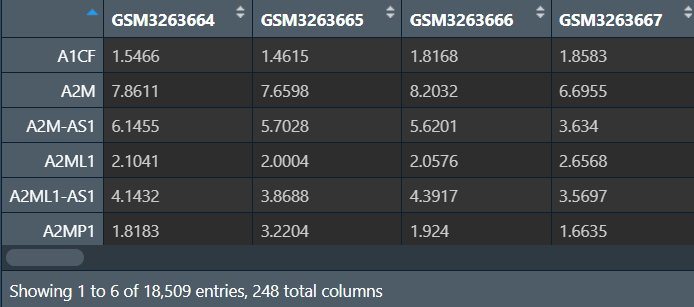
wd <- "data/GSE116918";
gset <- getGEO("GSE116918", destdir = wd, AnnotGPL = F, getGPL = F);
dat <- exprs(gset[[1]]);
get_log <- function(dat){
ex <- dat;
qx <- as.numeric(quantile(ex, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T));
LogC <- (qx[5]>100) || (qx[6]-qx[1]>50 && qx[2]>0) || (qx[2]>0 && qx[2]<1 && qx[4]>1 && qx[4]<2);
if(LogC){
ex[which(ex<=0)] <- NA;
ex <- log2(ex);
print("log2 transform finished");
}
else{
print("log2 transform not needed");
}
return(ex);
};
dat <- get_log(dat);
pd <- pData(gset[[1]]);
write.csv(pd, "data\\GSE116918\\clinical_GSE116918.csv");
读取soft文件:也是使用getGEO函数
注:soft文件也不用解压
GPL <- getGEO( # 读取soft文件
filename = "data\\GSE116918\\GPL25318_family.soft.gz",
destdir = wd
);
gpl <- GPL@dataTable@table; # 获取注释信息
只保留探针id和基因名
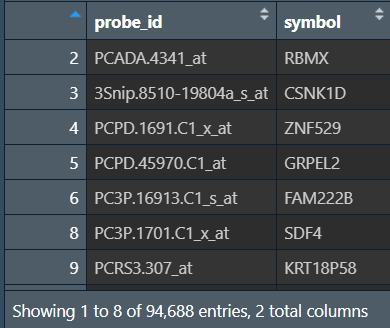
ids <- gpl[, c("ID", "Gene Symbol")]; # 保留探针id和基因名列
colnames(ids) <- c('probe_id', 'symbol'); # 改列名
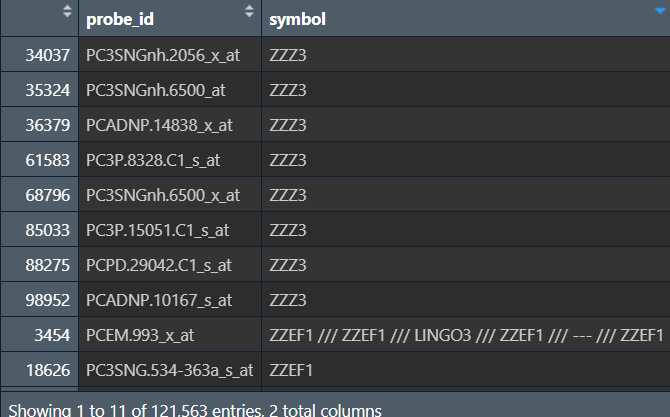
取出基因symbol
ids$symbol <- trimws(str_split(ids$symbol, "///", simplify = T)[, 1]); # 取出基因symbol
ids <- ids[ids$symbol != '', ];
ids <- ids[ids$symbol != '---', ]; # 去除空值
之后与第一种方法相同
ids <- ids[ids$probe_id %in% rownames(dat), ];
dat <- dat[ids$probe_id, ];
new_dat <- cbind(ids, dat);
new_dat <- aggregate(.~symbol, data=new_dat, FUN=max);
rownames(new_dat) <- new_dat[, 1];
new_dat <- new_dat[, -c(1, 2)];
write.table(data.frame(ID=rownames(new_dat),new_dat),
file = "data\\GSE116918\\GSE116918.txt", sep = '\t', quote = F, row.names = F);
代码汇总:
rm(list=ls());
if(!require("GEOquery", quietly = T))
{
BiocManager::install("limma");
BiocManager::install("GEOquery");
library("GEOquery");
}
if(!require("R.utils", quietly = T))
{
install.packages('R.utils');
library("R.utils");
}
library("tidyverse");
wd <- "data/GSE116918";
gset <- getGEO("GSE116918", destdir = wd, AnnotGPL = F, getGPL = F);
dat <- exprs(gset[[1]]);
get_log <- function(dat){
ex <- dat;
qx <- as.numeric(quantile(ex, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T));
LogC <- (qx[5]>100) || (qx[6]-qx[1]>50 && qx[2]>0) || (qx[2]>0 && qx[2]<1 && qx[4]>1 && qx[4]<2);
if(LogC){
ex[which(ex<=0)] <- NA;
ex <- log2(ex);
print("log2 transform finished");
}
else{
print("log2 transform not needed");
}
return(ex);
};
dat <- get_log(dat);
pd <- pData(gset[[1]]);
write.csv(pd, "data\\GSE116918\\clinical_GSE116918.csv");
GPL <- getGEO(filename = "data\\GSE116918\\GPL25318_family.soft.gz", destdir = wd);
gpl <- GPL@dataTable@table;
ids <- gpl[, c("ID", "Gene Symbol")];
colnames(ids) <- c('probe_id', 'symbol');
ids$symbol <- trimws(str_split(ids$symbol, "///", simplify = T)[, 1]);
ids <- ids[ids$symbol != '', ];
ids <- ids[ids$symbol != '---', ];
ids <- ids[ids$probe_id %in% rownames(dat), ];
dat <- dat[ids$probe_id, ];
new_dat <- cbind(ids, dat);
new_dat <- aggregate(.~symbol, data=new_dat, FUN=max);
rownames(new_dat) <- new_dat[, 1];
new_dat <- new_dat[, -c(1, 2)];
write.table(data.frame(ID=rownames(new_dat),new_dat),
file = "data\\GSE116918\\GSE116918.txt", sep = '\t', quote = F, row.names = F);
使用R包
对于一些较常使用的数据集,有人将注释信息封装到了R包中,数据集与R包对照
对于我们的数据,使用R包hgu133plus2.db
if(!require("hgu133plus2.db", quietly = T))
{
BiocManager::install("hgu133plus2.db");
library("hgu133plus2.db");
}
与第一种方式相同的地方不再说明
# 读取表达矩阵和临床信息部分同上
wd <- "data/GSE30219";
gset <- getGEO("GSE30219", destdir = wd, AnnotGPL = F, getGPL = F);
dat <- exprs(gset[[1]]);
get_log <- function(dat){
ex <- dat;
qx <- as.numeric(quantile(ex, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T));
LogC <- (qx[5]>100) || (qx[6]-qx[1]>50 && qx[2]>0) || (qx[2]>0 && qx[2]<1 && qx[4]>1 && qx[4]<2);
if(LogC){
ex[which(ex<=0)] <- NA;
ex <- log2(ex);
print("log2 transform finished");
}
else{
print("log2 transform not needed");
}
return(ex);
};
dat <- get_log(dat);
pd <- pData(gset[[1]]);
write.csv(pd, "data\\GSE30219\\clinical_GSE30219.csv");
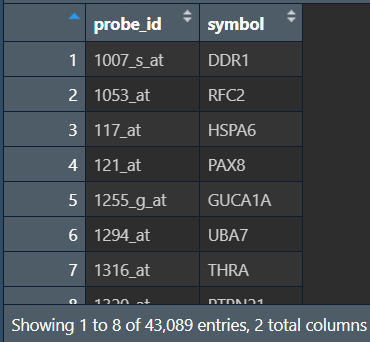
获取该包中的symbol注释信息:
ids <- toTable(hgu133plus2SYMBOL); # 获取注释信息
colnames(ids) <- c('probe_id', 'symbol'); # 改列名
因为是写好的包,无需再字符串处理
之后与前面方法相同
ids <- ids[ids$probe_id %in% rownames(dat), ];
dat <- dat[ids$probe_id, ];
new_dat <- cbind(ids, dat);
new_dat <- aggregate(.~symbol, data=new_dat, FUN=max);
rownames(new_dat) <- new_dat[, 1];
new_dat <- new_dat[, -c(1, 2)];
write.table(data.frame(ID=rownames(new_dat),new_dat),
file = "data\\GSE30219\\GSE30219.txt", sep = '\t', quote = F, row.names = F);
代码汇总:
rm(list=ls());
if(!require("GEOquery", quietly = T))
{
BiocManager::install("limma");
BiocManager::install("GEOquery");
library("GEOquery");
}
if(!require("R.utils", quietly = T))
{
install.packages('R.utils');
library("R.utils");
}
if(!require("hgu133plus2.db", quietly = T))
{
BiocManager::install("hgu133plus2.db");
library("hgu133plus2.db");
}
library("tidyverse");
wd <- "data/GSE30219";
gset <- getGEO("GSE30219", destdir = wd, AnnotGPL = F, getGPL = F);
dat <- exprs(gset[[1]]);
get_log <- function(dat){
ex <- dat;
qx <- as.numeric(quantile(ex, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T));
LogC <- (qx[5]>100) || (qx[6]-qx[1]>50 && qx[2]>0) || (qx[2]>0 && qx[2]<1 && qx[4]>1 && qx[4]<2);
if(LogC){
ex[which(ex<=0)] <- NA;
ex <- log2(ex);
print("log2 transform finished");
}
else{
print("log2 transform not needed");
}
return(ex);
};
dat <- get_log(dat);
pd <- pData(gset[[1]]);
write.csv(pd, "data\\GSE30219\\clinical_GSE30219.csv");
ids <- toTable(hgu133plus2SYMBOL);
colnames(ids) <- c('probe_id', 'symbol');
ids <- ids[ids$probe_id %in% rownames(dat), ];
dat <- dat[ids$probe_id, ];
new_dat <- cbind(ids, dat);
new_dat <- aggregate(.~symbol, data=new_dat, FUN=max);
rownames(new_dat) <- new_dat[, 1];
new_dat <- new_dat[, -c(1, 2)];
write.table(data.frame(ID=rownames(new_dat),new_dat),
file = "data\\GSE30219\\GSE30219.txt", sep = '\t', quote = F, row.names = F);
TCGA与GEO多数据集的合并及去除批次效应
使用包:sva、limma
if(!require("sva", quietly = T))
{
library("BiocManager");
BiocManager::install("sva");
library("sva");
}
if(!require("limma", quietly = T))
{
library("BiocManager");
BiocManager::install("limma");
library("limma");
}
使用的数据:
-
TCGA数据集
TCGA_LUSC_TPM.txt -
GEO数据集
GSE74777.txt和GSE30219.txt
读取数据:
-
转化为matrix格式
-
对TCGA数据取log2
-
对GEO数据进行标准化
my_read <- function(path){
data <- read.table( # 读取数据
file = path,
header = T,
sep = '\t',
check.names = F,
row.names = 1
);
dimnames <- list(rownames(data), colnames(data)); # 提取行名列名
data <- matrix( # 转为矩阵
as.numeric(as.matrix(data)), # 转为数值型
nrow = nrow(data), # 行数
dimnames = dimnames # 指定行名列名
);
return(data);
}
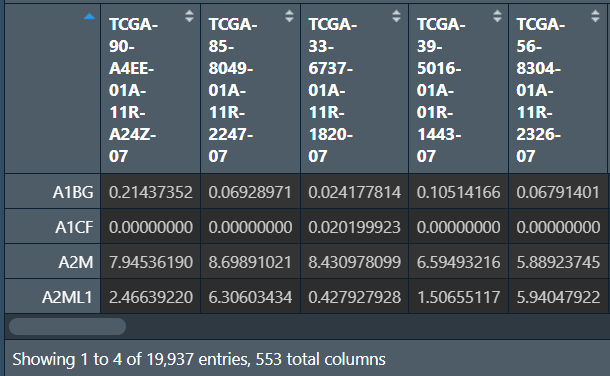
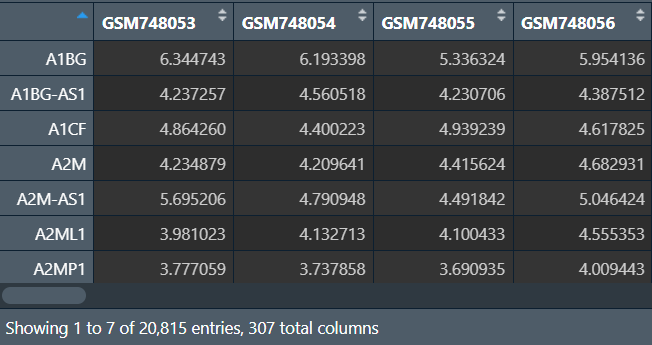
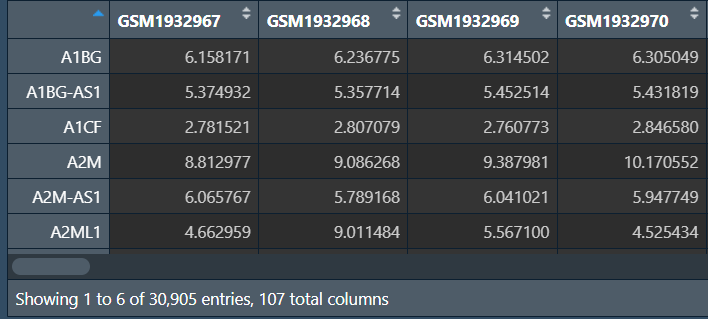
rt1 <- my_read("data\\merge\\TCGA_LUSC_TPM.txt");
rt2 <- my_read("data\\merge\\GSE30219.txt");
rt3 <- my_read("data\\merge\\GSE74777.txt");
rt1 <- log2(rt1+1); # 为防止有0值,所以+1
rt2 <- normalizeBetweenArrays(rt2);
rt3 <- normalizeBetweenArrays(rt3);
合并:
-
获取三数据集中共同的基因
-
对三数据集中共有基因进行列合并
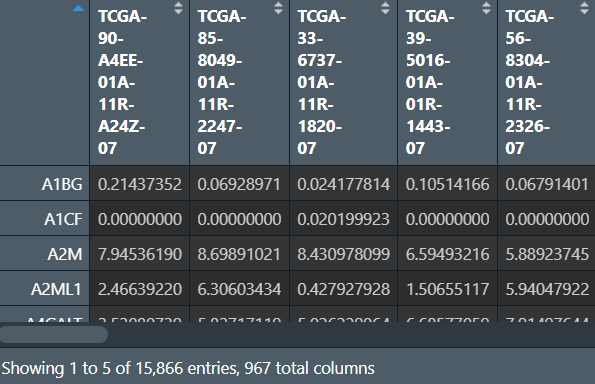
gene <- intersect(rownames(rt1), rownames(rt2));
gene <- intersect(gene, rownames(rt3)); # 共有基因
merge_data <- data.frame(); # 初始化结果矩阵
merge_data <- cbind(rt1[gene, ], rt2[gene, ]);
merge_data <- cbind(merge_data, rt3[gene, ]); # 合并
去除批次效应:
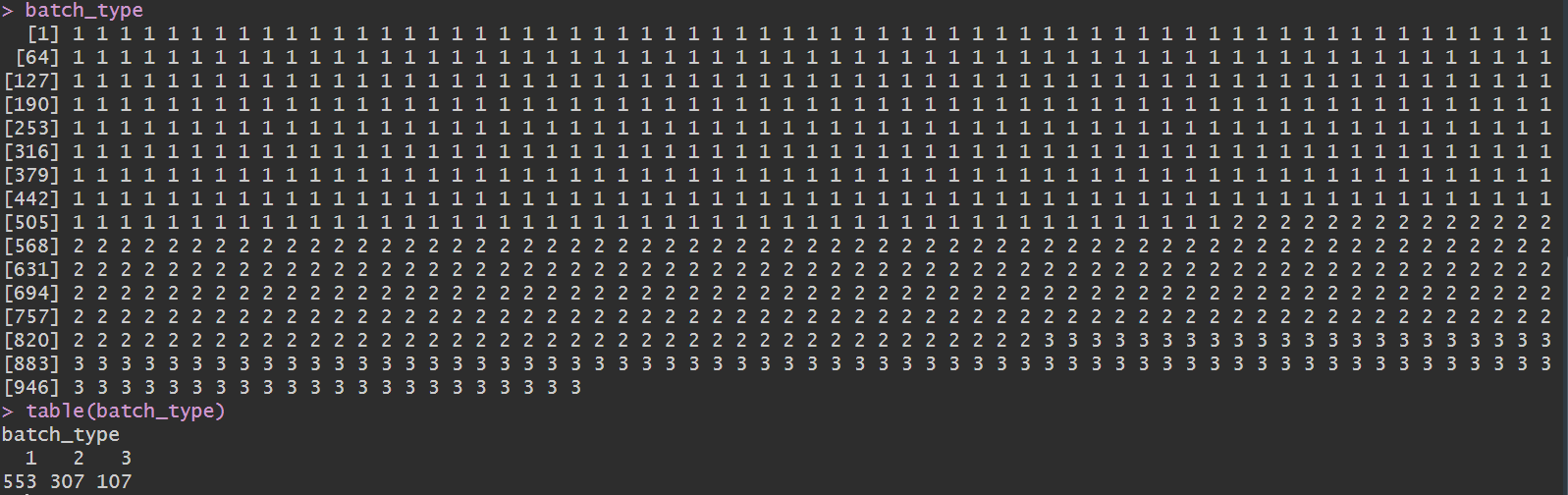
创建分组信息,标识merge_data的第几列属于哪组数据
# 使用rep(x, time),表示将x复制time次
batch_type <- c();
batch_type <- c(batch_type, rep(1, ncol(rt1)));
batch_type <- c(batch_type, rep(2, ncol(rt2)));
batch_type <- c(batch_type, rep(3, ncol(rt3)));
使用ComBat函数进行批次矫正
outTab <- ComBat(merge_data, batch_type, par.prior = T);
save_outTab <- rbind(
ID = colnames(outTab),
outTab
);
write.table(
save_outTab,
file = "save_data\\merge.txt",
sep = '\t',
quote = F,
col.names = F
);
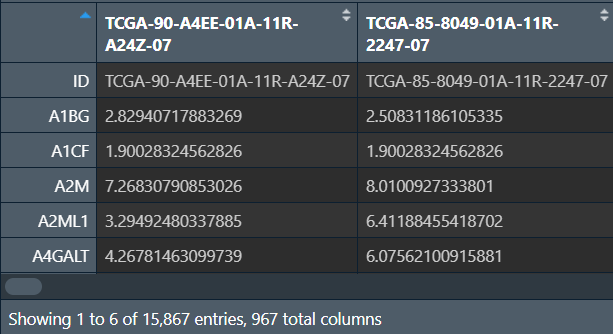
行名是基因名,列名是样本名,数据值是批次校正后的表达矩阵
可以将上述代码进行封装,使其能适用于任意数量的数据合并(推荐使用)
files <- c("data\\merge\\TCGA_LUSC_TPM.txt", "data\\merge\\GSE30219.txt", "data\\merge\\GSE74777.txt"); # 需要合并的数据
gene_list <- list(); # 元素是数据矩阵
# 读取数据并初步处理
for (i in 1:length(files)) {
rt <- read.table(files[i], header = T, sep = '\t', check.names = F, row.names = 1); # 读取数据
dimnames <- list(rownames(rt), colnames(rt)); # 提取行名列名
rt <- matrix(as.numeric(as.matrix(rt)), nrow = nrow(rt), dimnames = dimnames); # 转为矩阵
if(substr(colnames(rt)[1], 1, 3)=='TCG'){ # 如果是TCGA数据
rt <- log2(rt+1); # 就取log2
}
if(substr(colnames(rt)[1], 1, 3)=='GSM'){ # 如果是GEO数据
rt <- normalizeBetweenArrays(rt); # 就标准化
}
gene_list[[i]] <- rt; # 添加到结果列表中
# 取共同基因
if(i==1){ # 第一次循环给gene赋初值
gene <- rownames(gene_list[[1]]);
}
else{ # 否则取交集
gene <- intersect(gene, rownames(gene_list[[i]]));
}
}
# 合并并创建分组信息
for (i in 1:length(files)) {
if(i==1){ # 第一次循环,给结果矩阵和分组信息赋初值
merge_data <- gene_list[[1]][gene, ];
batch_type <- c(rep(1, ncol(gene_list[[1]])));
}
else{ # 否则进行合并
merge_data <- cbind(merge_data, gene_list[[i]][gene, ]);
batch_type <- c(batch_type, rep(i, ncol(gene_list[[i]])));
}
}
# 去除批次效应
outTab <- ComBat(merge_data, batch_type, par.prior = T);
save_outTab <- rbind(ID = colnames(outTab), outTab);
write.table(save_outTab, file = "save_data\\merge2.txt", sep = '\t', quote = F, col.names = F);