b站生信课程01
r-bioinfolessonb站生信课程生信从0到1学习系列笔记
写在前面:本篇教程来自b站课程生信从0到1学习系列
课程资料 提取码:tjzg
测序相关概念
-
read:测序过程中要将一个基因切分成多个小段DNA,每对小段进行一次测序,就称为一次read
-
count:一个基因所有read的总数
在某样品中,基因A的count>基因B,但这不能说明基因A的表达一定比B高,可能只是因为基因A更长
在两个样品中,其中一个样品基因A的count比另一个样品高,也不能说明这个样品基因A表达更多,可能是因为PCR时这个样品基因A扩增次数更多
-
转录本(transcript):基因转录成pre-mRNA后,因为基因有内含子和外显子,需要对pre-mRNA进行剪切,有不同的剪切方法,产生不同的转录本
-
基因长度:有多少个碱基对(不包括内含子),大概有4种定义方式
-
基因最长的转录本长度
-
多个转录本长度的平均值
-
非重叠外显子长度和(L1+L2+L3+L4)–常用
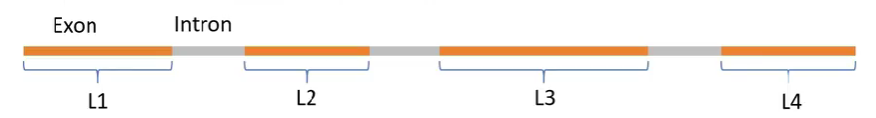
-
非重叠编码序列长度和
-
-
测序深度:测序得到的碱基总量(bp)与基因组(或转录组、测序目标区域)大小的比值
简单理解:PCR扩增后,DNA片段越多,测序的时候得到的碱基总量也就越多,测序深度越大
标准化
-
RPK:
count/基因长度*103,即每千个碱基的read数用于同一个样品中,比较不同基因表达量,可以减少基因长度的影响
-
RPKM/FPKM:某个样品中
某基因的count值/该基因长度/所有基因count值之和*109,即每千个转录、每百万映射读取的read数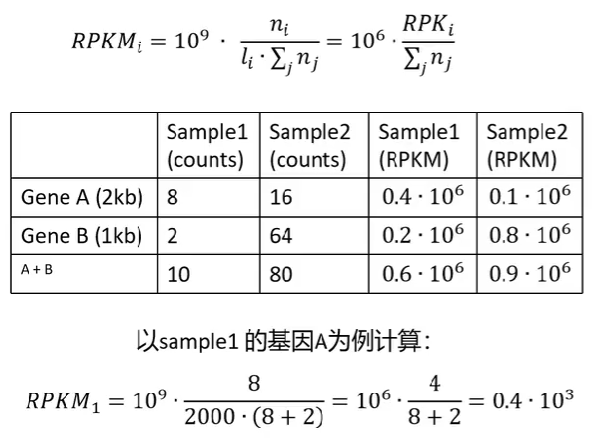
在减少基因长度的影响后,再减少测序深度的影响
单端测序使用RPKM,双端测序(得到正反两个测序结果)使用FPKM
可以在样品内比较,无法在样品间比较。以上图为例,只能说明样品1中基因A表达量高于B,不能说明样品1中基因A表达量高于样品2中基因A表达量
-
TPM:某个样品中
某基因的count值/该基因长度/所有基因count值与其基因长度比值之和*106,即每千个转录、每百万映射读取的transcripts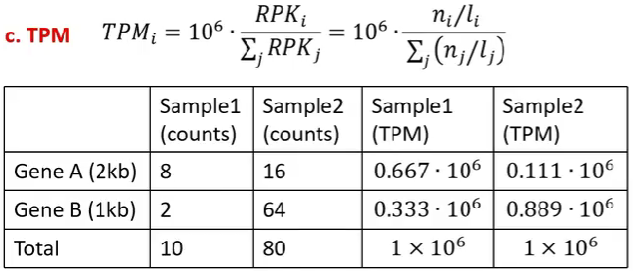

既能组内比较,又能组间比较
-
CPM(使用较少):某个样品中
某基因的count值/所有基因的count值之和*106,即每百万映射读取的reads
可以用于组件比较,不能用于组内比较
在使用R包作差异分析时,有些R包(如DESeq2)只能输入count值,而不能输入标准化后的值,因为它有自己的一套标准化算法,
总结:

常用分析方法
以用于头颈部鳞状细胞癌患者生存、治疗结果、免疫浸润的ADME相关基因签名的探索和验证论文为例
ADME:药物在体内的吸收(Absorption)、分布(Distribution)、代谢(Metabolism)、和排泄(Excretion),属于药代动力学研究的内容
差异分析
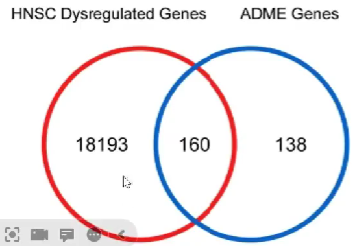
通过肿瘤与正常细胞基因表达的差异,筛选出18353个差异表达基因
再将18353个差异表达基因与298个ADME基因取交集,得到160个差异表达的ADME基因

单因素cox分析
通俗来讲,就是为了研究哪类群体的“死亡”速度更快、什么因素影响了“死亡”速度
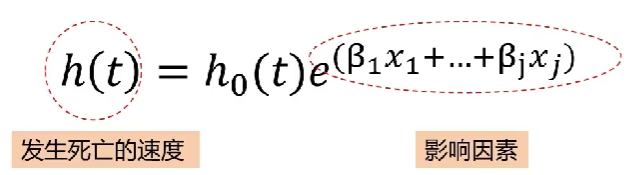 其中h0(t)为常数
其中h0(t)为常数
两个简单的例子:
-
想知道性别对某疾病的影响,即是男性死亡风险高,还是女性死亡风险高。可以在t时刻,用女性的死亡风险除以男性的死亡风险,最后得到一个风险比
HR,当其大于1且有统计学意义时就可以说女性死亡风险更高 -
想知道多个基因对死亡的影响,可以分别计算它们的风险比
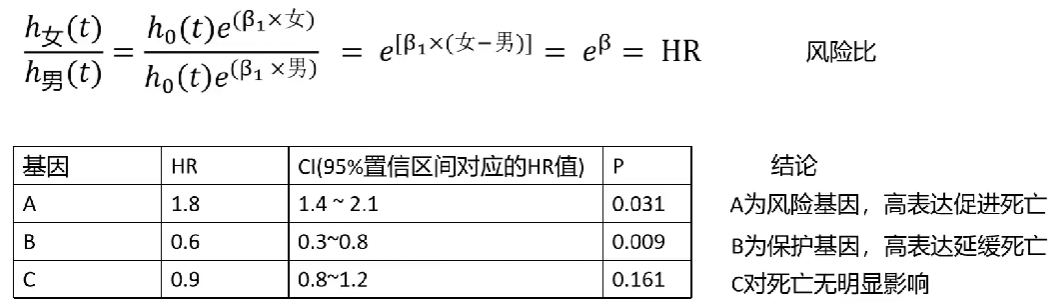
无需记住公式,R包会提供函数进行计算
在实际应用中,会在很多个基因中进行筛选,如
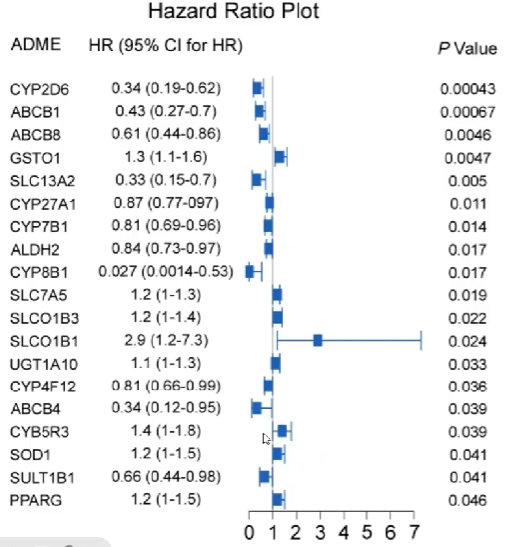
一共得到了19个有价值的差异表达基因,HR小于1为保护基因,大于1为风险基因
生存曲线
如何验证通过单因素cox分析得到的基因是否真的有价值?
将每个基因根据表达量的中位数/均值,分为高表达与低表达两组,绘制曲线
曲线的横坐标为时间,纵坐标为生存概率
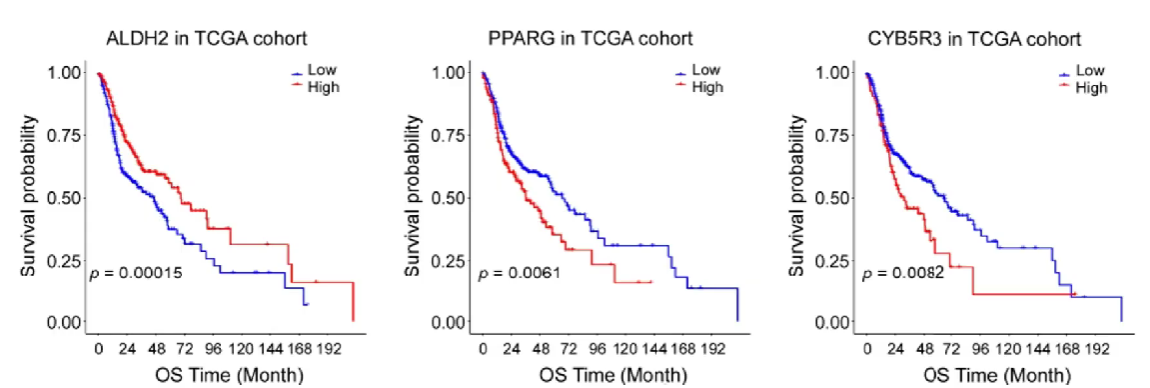
比较同一时刻高/低表达组的生存概率,就可以知道单因素cox分析预测的是否准确
分子互作网络
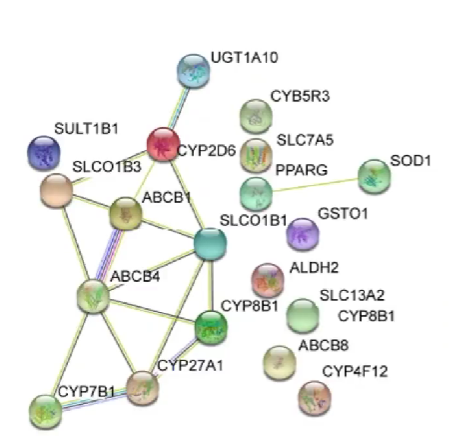
-
圆圈:即旁边标注的基因
-
圈里有螺旋结构:代表这个基因的分子结构已知,没有则代表未知
-
圆圈颜色:不同颜色代表不同功能的基因
-
线条:表示基因之间存在的联系,没有则代表现在还不知道是否有联系
-
线条颜色:代表分子间不同的关系,具体可以在string官网 中找到
LASSO-COX筛选基因构建模型
LASSO回归
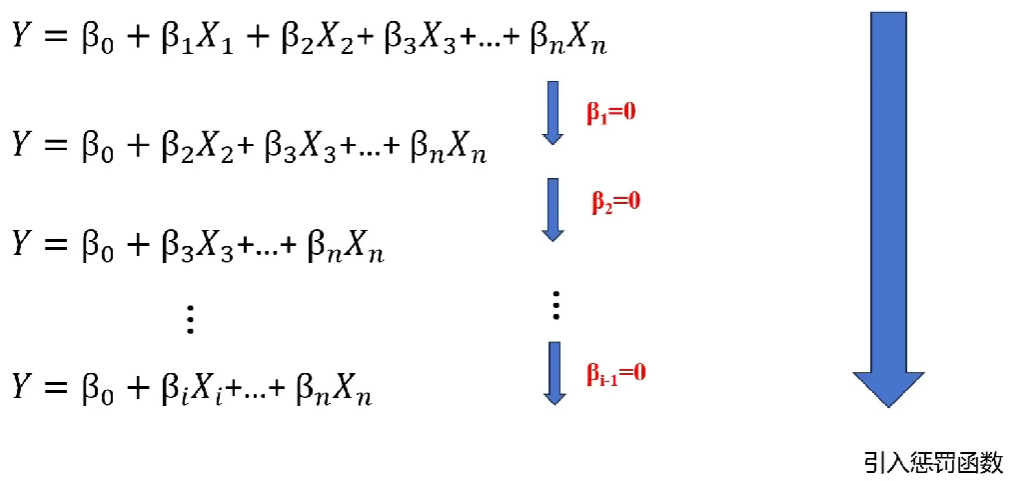
-
等号右边为n个影响因素及权重,X为影响因素,β为常数(代表对应X的权重)
-
等号左边的Y可以称为受它们影响的结果
在这些影响因素中,有些对结果的影响权重较高,有些较低。可以剔除一些影响因子,同时让Y不发生太大的改变,保留权值高的影响因子
LASSO回归提供了一个惩罚函数,逐渐让权值低的X的β=0,最后只留下必要的影响因子,从而简化模型
回归系数路径图:
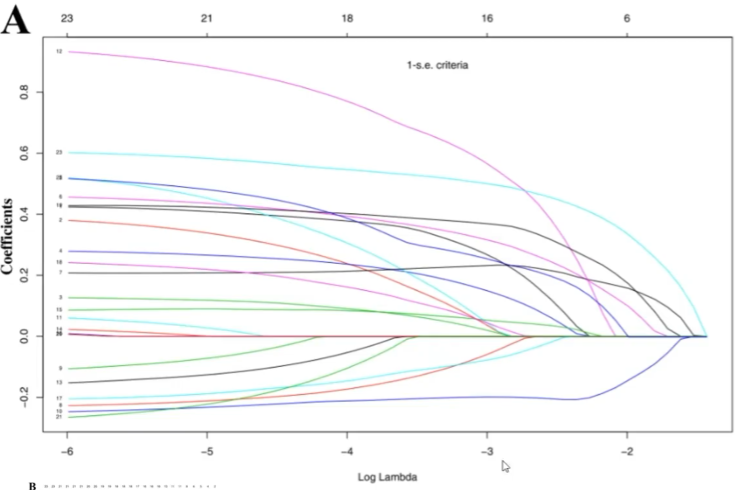
-
纵轴:回归系数
-
横轴:log λ(LASSO回归中的一个参数)
-
多少条线就代表了多少个变量的回归系数
-
最上面的一排数字表示取对应的log(λ)时,还有多少个基因的系数不为0,下同
随着λ不断变大,越来越多的回归系数收敛到0,回归方程越来越简单,最后只剩下Y=β0,这显然是不合理的,因此需要知道当λ为多少时才能得到合理的结果(保留多少个影响因素)
交叉验证曲线:
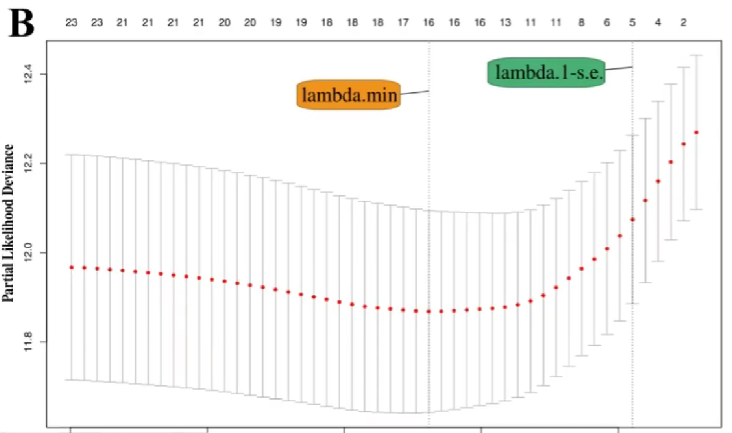
-
纵轴:似然偏差,越小代表拟合效果越好
-
横轴:log λ(同回归系数路径图)
-
λ min:偏差最小时的log λ值,此时模型的拟合效果最好 -
λ1-se(λ1减se):表示λ min右侧的一个标准误,此时模型的拟合效果也很好,同时模型的变量更少、模型更简单 -
很多时候以λ1-se作为筛选标准,实际上
λ min~λ1-se范围内都可以
通过LASSO回归,在上面的19个基因中筛选了其中14个关键基因
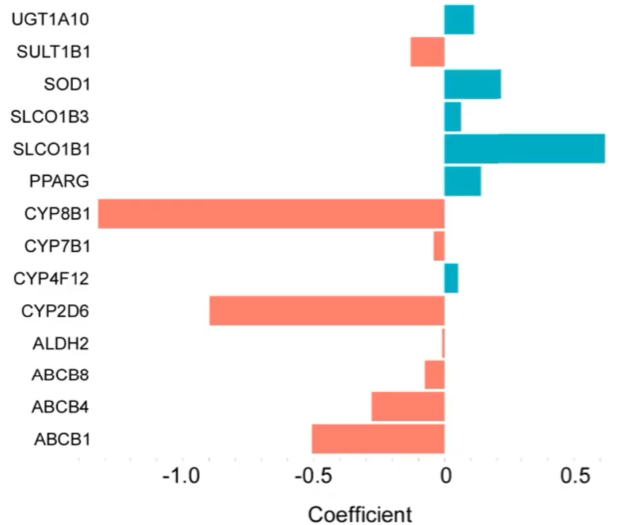
去掉了其中4个影响权重较低的基因,同时保证了模型预测效果
生存分析与ROC曲线
在完成上面的步骤后,根据14个关键基因在每个样本中的表达值,对每个样品进行风险打分。此例中共有274个样本,因此有274个风险得分
之后根据风险得分将这些样本分为高风险组和低风险组(设置分组阈值为风险得分的中位数)
对高/低风险组进行生存分析:
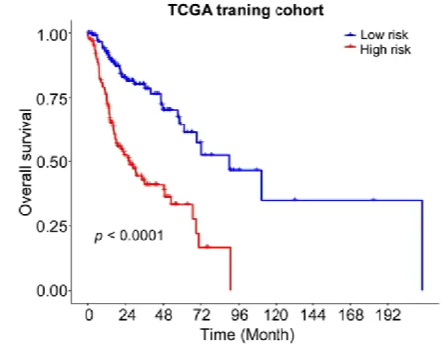
并绘制ROC曲线:
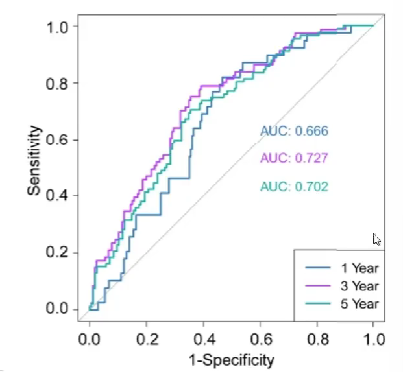
在ROC曲线中,只需关注AUC值,上图中对角线(灰色)即为AUC=0.5的基准线
-
ROC曲线接近左上角(AUC值较大):模型预测准确率高
-
ROC曲线略高于基准线(AUC值略大于0.5):模型预测准确率一般
-
ROC曲线低于基准线(AUC值小于0.5):模型未达到最低标准,无法使用
模型验证
内/外部验证
-
内部验证:将最开始建模所用到的数据集分成两部分,一部分用于建模,另一部分用于验证
-
外部验证:用另外一批测序的数据
重复上面的步骤:计算验证组风险得分,根据之前算出的分组阈值将它们分为高风险组和低风险组,画出生存分析图和ROC曲线
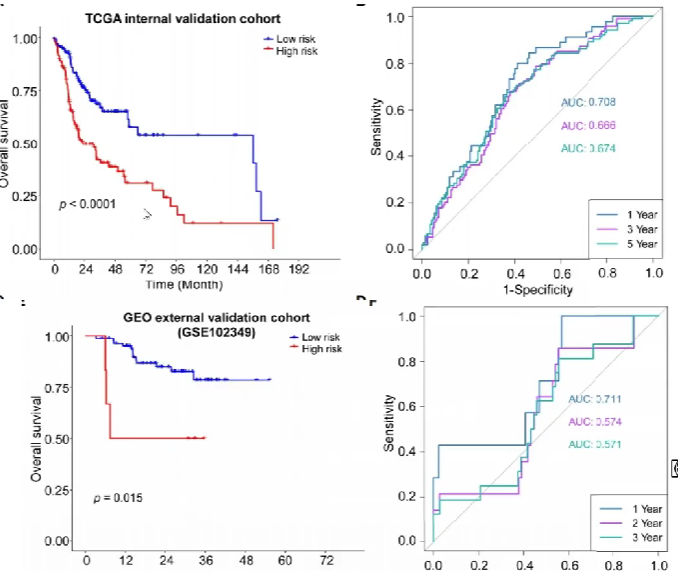
可以看到内部验证效果较好(因为都来自同一批数据),外部验证中1year的效果较好,2year和3year效果一般
独立性验证
即探究某个因素对预测结果的影响
此例中探究了淋巴血管是否浸润对结果的影响
按淋巴血管是否浸润分为有浸润组和无浸润组,对这两组分别重复上面的步骤(分为高风险组和低风险组,画出生存分析图),得到两张生存分析图
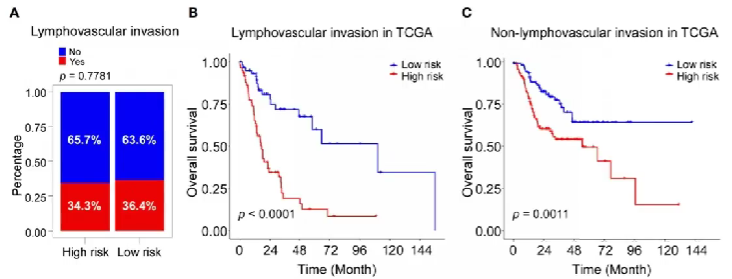
在这两组中高风险组生存率都低于低风险组,且低/高风险组中出现浸润的病人比例类似
得出结论:生存分析结果不受淋巴血管是否浸润的影响
GO与KEGG富集分析
统计各差异表达基因的生物学功能
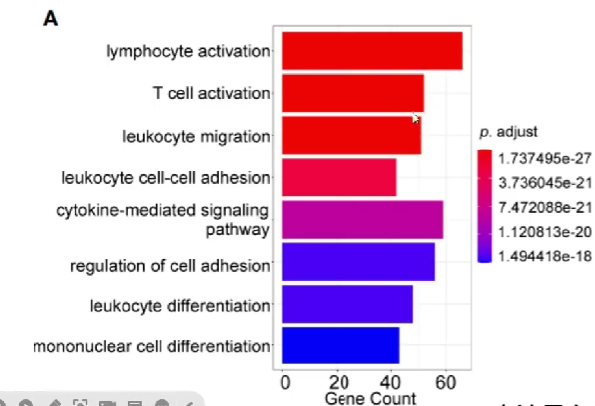
纵轴为生物学功能,横轴为具有某项生物学功能的基因总数
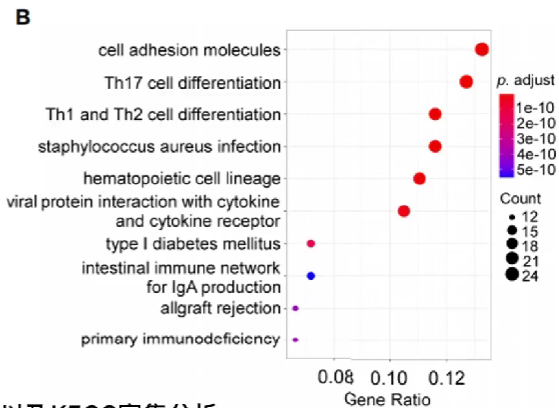
纵轴为生物学功能,横轴为具有某项生物学功能的基因比例
可以看到它们的p值很小,说明有统计学意义
此例中大多数基因都与免疫学功能有关,因此需要进行免疫分析
免疫分析
为了弄清肿瘤组织中免疫细胞的构成比(免疫微环境),其对肿瘤的发展、治疗有很大作用
主要有2种方法:
-
单细胞测序,得到各个细胞亚群比例。缺点是成本很高
-
根据RNA-seq结果推测处组织中各免疫细胞构成
对高/低风险组分别进行免疫分析,并汇总到一个图上,可以得到各种免疫细胞在高/低风险组中的数量差别
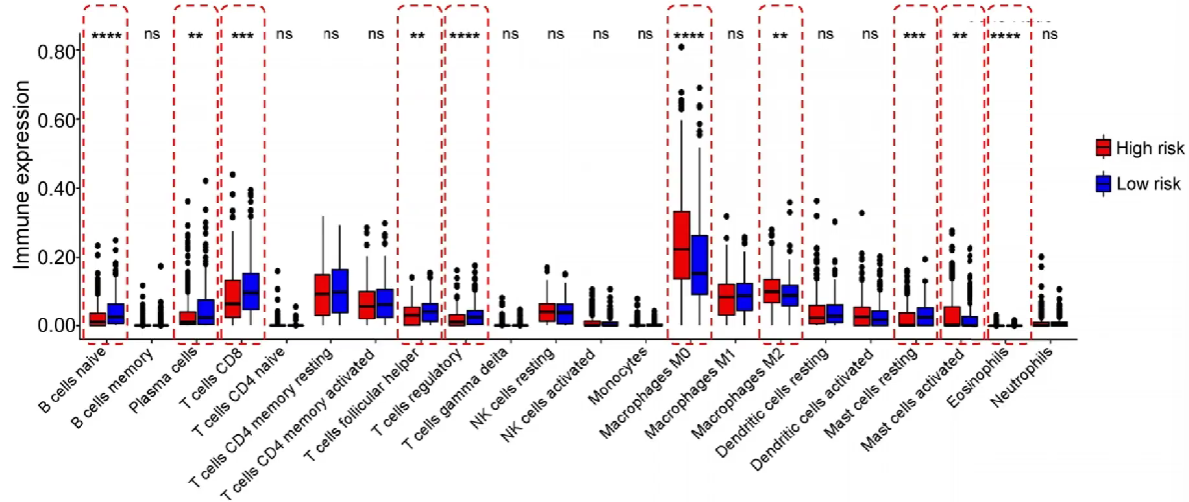
共统计了22种免疫细胞,其中10种(红色虚线部分)在高/低风险组中的比例有显著差异
免疫评分:将上面得到的有显著差异的免疫细胞单独进行风险评分
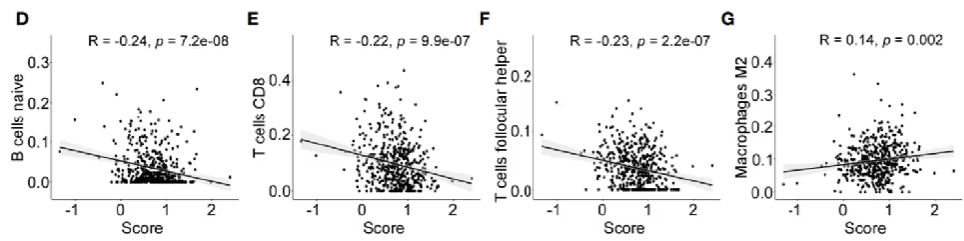
图中的拟合曲线表明了该种免疫细胞对风险影响:斜率<0则有利,斜率>0则有害
三种评分:
-
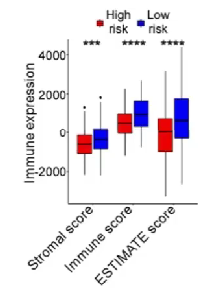
Stromal score(基质评分):评估肿瘤组织中的基质细胞浸润水平
-
Immune score(免疫评分):评估肿瘤组织中的免疫细胞浸润水平
-
Estimate Score(可推测帅瘤纯度) = stromal score + immune score
上述评分越高,肿瘤纯度越低,预后越好
治疗反应
即高/低风险组经过免疫治疗/化疗后的生存分析
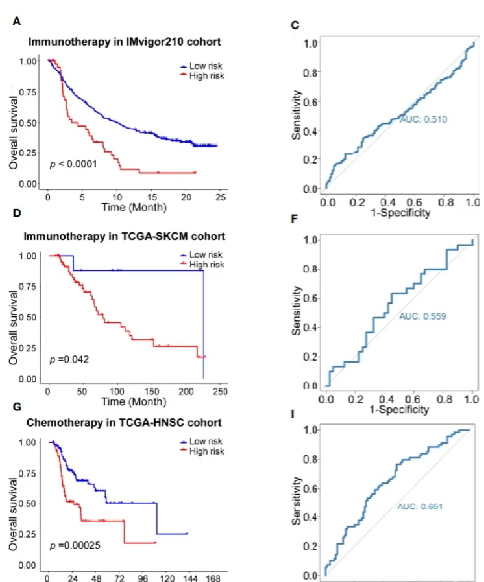
可以看到免疫治疗组预测效果较差,化疗组预测效果较好
总结
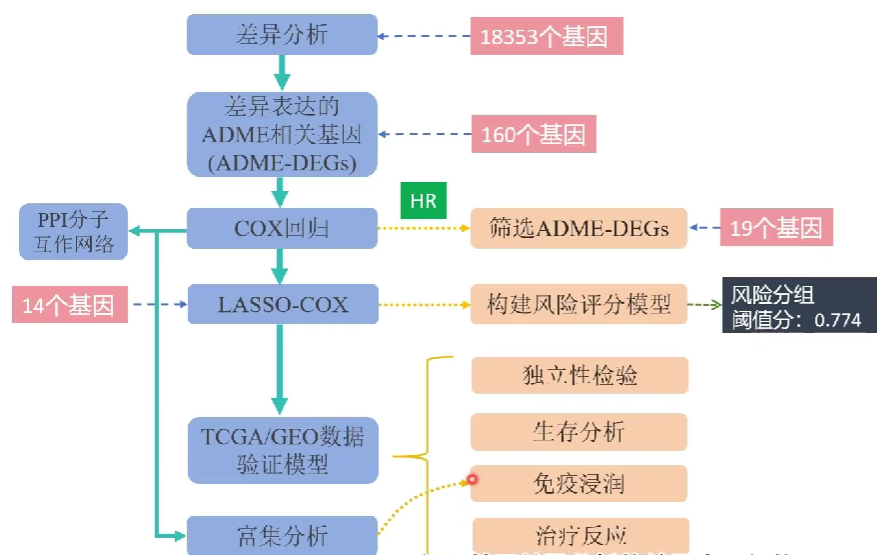
简化一下:
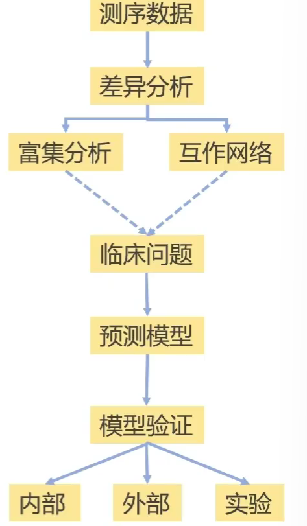
-
差异分析是基础,它得到要进行分析的基因
-
富集分析让我们知道差异基因的哪项功能多,方便后续针对性的研究
-
互作网络得到分子间的相互作用
-
根据具体的临床问题构建相应的模型,并进行验证(内部、外部、实验验证)
为什么要实验验证?
基因表达,蛋白质不一定表达。而真正影响身体的是表达出的蛋白质,需要通过实验检测蛋白质的表达
常用数据库
GEO数据库
GEO数据库:由NCBI创建并维护的高通量基因表达数据库
有GEO Datasets和GEO Profiles两个子数据库:
-
GEO Datasets:以基因为单位,存储基因在数据集中的表达谱
-
GEO DataSets:以一次完整实验为单位,收录整个试验的数据集,是一个试验中的完整数据集
-
Platform:平台编号,以”GPL”开头
-
Samples:样本编号,以”GSM”开头
-
Series:将构成某个试验的相关数据集中到一个完整的数据集中,包括实验设计、描述、组别、样本等信息以及检测数据文件,以”GSE”开头
-
在下载GEO数据的过程中,最重要的就是Series
使用
以头颈部鳞状细胞癌(head and neck cancer)为例:
进入GEO官网
第一种方法:
搜索head and neck cancer
点击There are 18010 results for "head and neck cancer" in the GEO DataSets Database
如果想以人类作为研究对象,就在右上角organisms中选择Homo sapiens
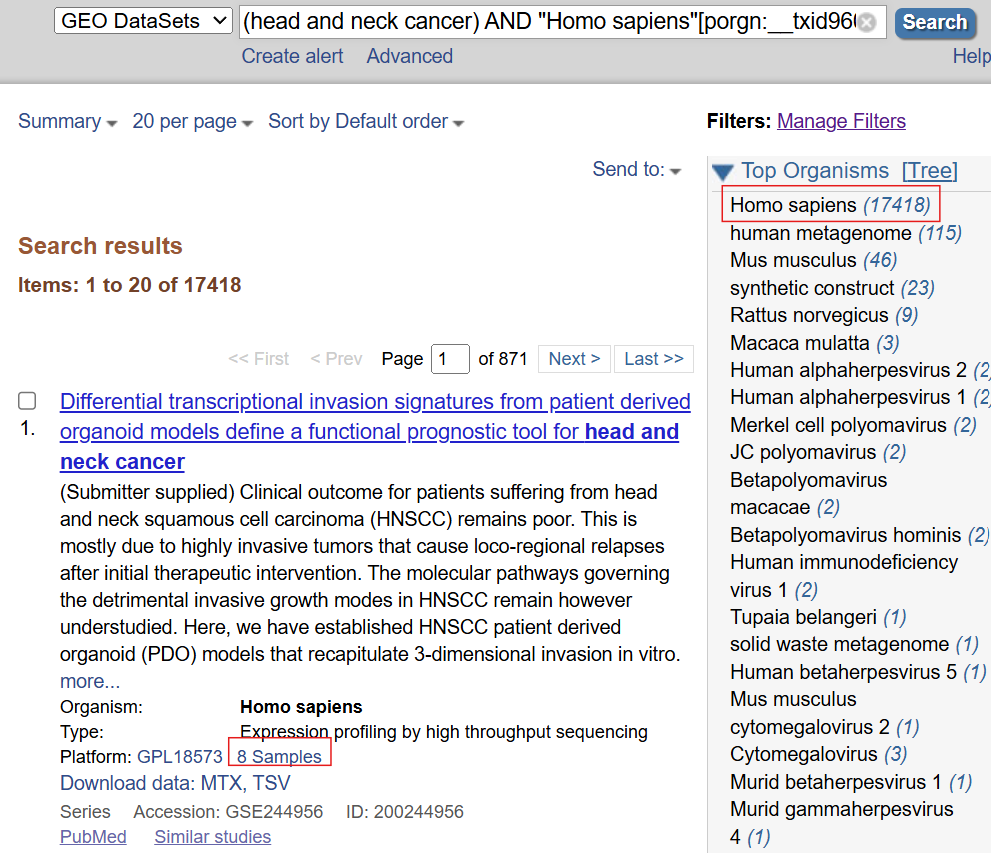
在每个标签内可以看到样本数,样本数过小的数据不能用于分析,需要将多个小样本数的数据合并。这里为了简单,直接过滤掉小样本数的数据,找样本足够多的数据:
-
在左侧栏中找到
Show additional filters,勾选Sample count,点击show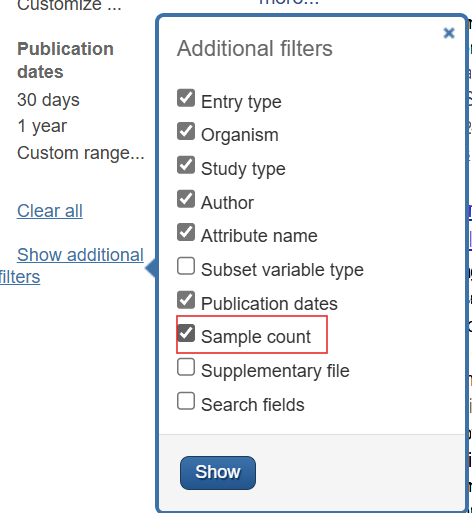
-
再在它上面选择
Sample countCustom range设置范围>30,点击apply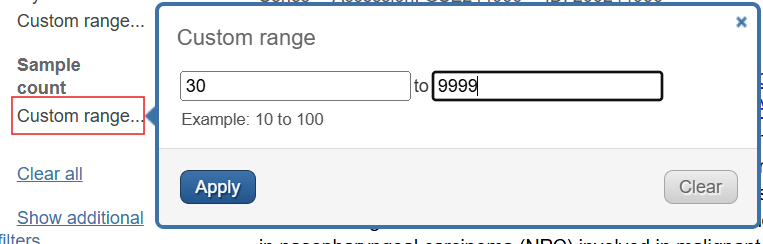
发现此时仍有数百条数据,且很多都不是关于头颈部鳞状细胞癌的
第二种方法:更加精准
点击首页中Browse Content的Series
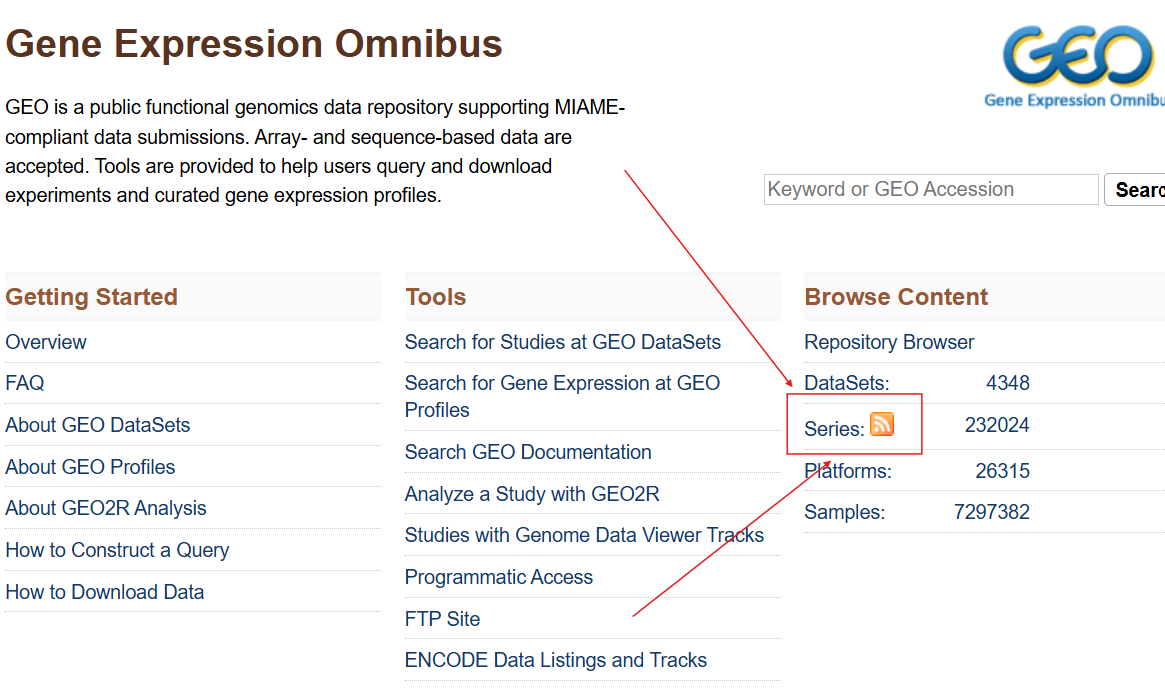
在打开的界面中搜索head and neck cancer
可以看到只有100条左右的数据
直接点击organisms列中的Homo sapiens就可以筛选出人类的数据
点击samples列就可以按样本数从高到低排序
以GSE65858基因集为例,点击GSE65858链接即可进入
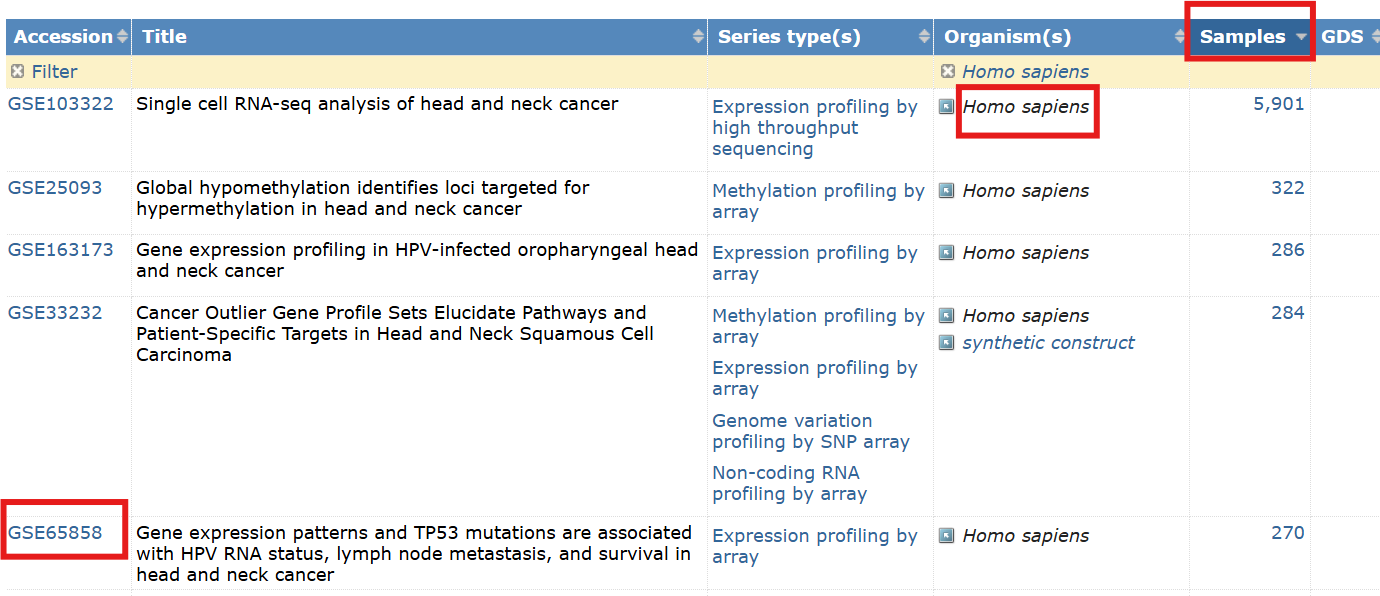
其中有
-
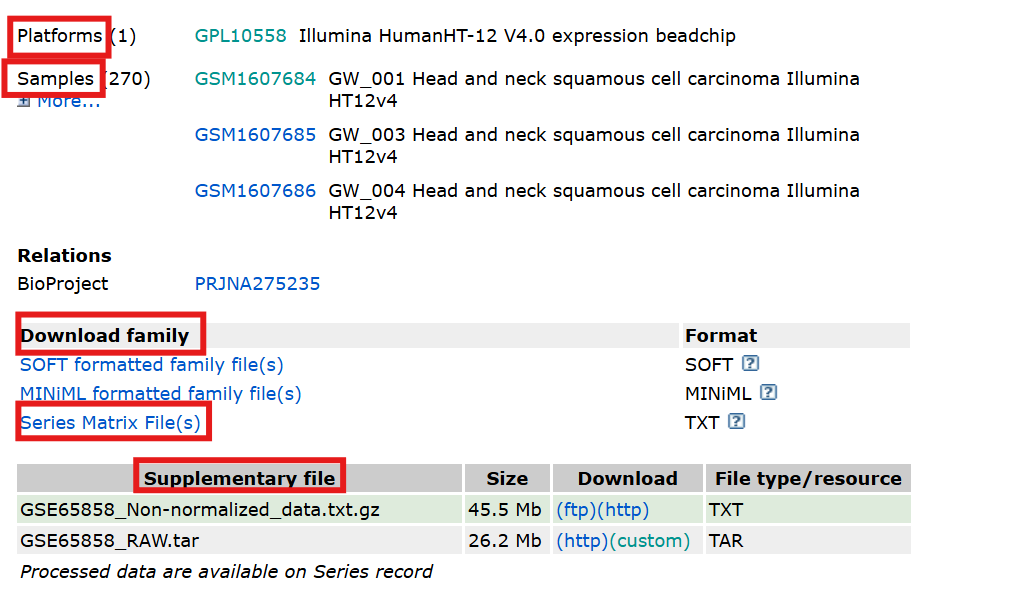
测序平台信息
Platforms点进去,重点看
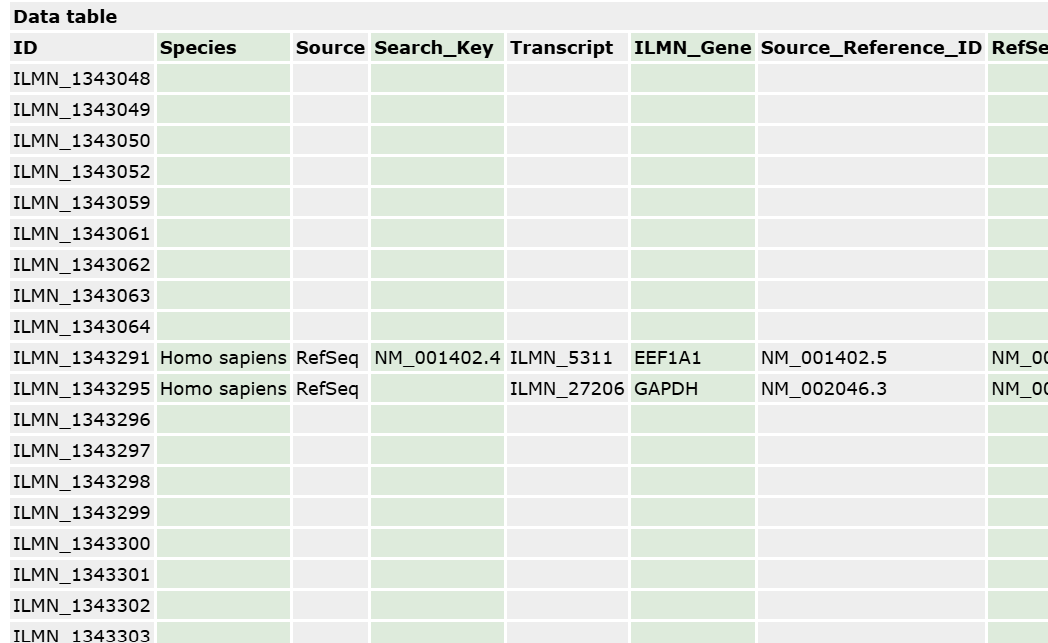
data table第一列是基因id,我们下载完测序数据后,需要和这个基因id对应起来,找到基因名称等
-
样本信息
Samples其中
os/os_time表示生存时间(从确诊到最近一次随访),os_event表示阳性事件是否发生(1为发生、0为不发生),这里的阳性事件指是否死亡 -
下载原始测序数据
Supplementary file样本中的数据一般都是将counts标准化后的
-
Non-normalized_data就是未标准化的数据 -
RAW就是最原始的测序数据,未计算counts
-
-
下载数据集
Download family其中最关注的就是
Series Matrix File(s)点进去后可以看到文件大小,一般数量级是M,说明包含了表达、临床信息等
如果是KB级,就需要到
Supplementary file中找表达数据等
使用R下载GEO数据
下载GEOquery包:参考文章
(如不存在该网址就到bioconductor官网中手动搜索GEOquery)
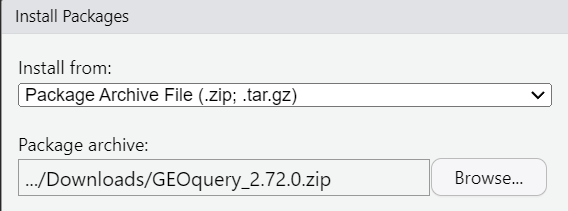
找到Package Archives部分,根据系统选择相应版本,一般用zip文件
在rstudio中点击上面的Tools->install packages,install from选择package archive file,选择刚刚下载的压缩包,即可安装
也可使用
install.packages("BiocManager")
library("BiocManager");
BiocManager::install("GEOquery")
之后使用GEOquery包中的getGEO函数进行下载
geo_set <- getGEO(
GEO='数据集名称', # 即GSE开头的数据集编号
filename='已下载的数据集地址', # 读取已下载的数据集,注意该参数与GEO=只能指定一个
destdir='目标文件夹', # 要下载到哪个文件夹
GSElimits=NULL, # 指定GSE子集,需要下载哪些样本
GSEMatrix=TRUE, # 是否需要下载matrix文件
AnnotGPL=FALSE, # 是否使用Annotation GPL(注释基因的文件),很多数据集没有这项
getGPL=FALSE # 是否下载Annotation GPL,用于注释基因
)
具体使用:
geo_set <- getGEO(
GEO = "GSE65858",
destdir = '.', # 表示下载到当前工作目录
AnnotGPL = F,
getGPL = F
);
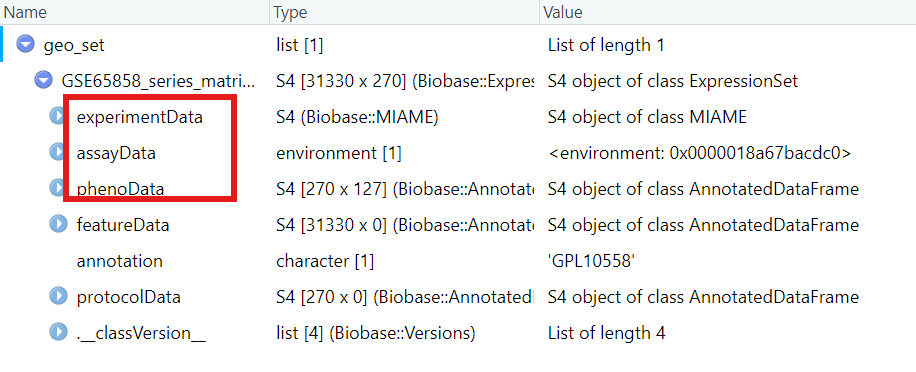
查看下载得到的geo_set:
其中
-
experimentData是实验信息,包括姓名、实验室、联系方式、摘要等等
-
assayData是表达信息
-
phenoData是临床信息
常用的两个函数:
expr <- exprs(geo_set[[1]]);
把表达信息复制到expr变量中,列名是样本名称,行名是基因id
有时下载得到的数据中没有表达数据,得到的expr为空,这时就需要到GEO网站上下载原始数据进行进一步分析
pd <- pData(geo_set[[1]]);
得到临床信息pd,行名是样本名称,列名包括肿瘤信息、os、os_event等等

提示:有时使用已下载的数据集进行getGEO(),得到的数据用这两个函数处理时可能报错函数...标签...找不到继承方法
虽然从网站上下载的数据与getGEO()下载得到的数据内容相同,但外层格式会有一定差别:
geo_set <- getGEO(
filename = "GSE65858_series_matrix.txt.gz",
destdir = '.',
AnnotGPL = F,
getGPL = F
);
geo_set2 <- getGEO(
GEO = "GSE65858",
destdir = '.',
AnnotGPL = F,
getGPL = F
);
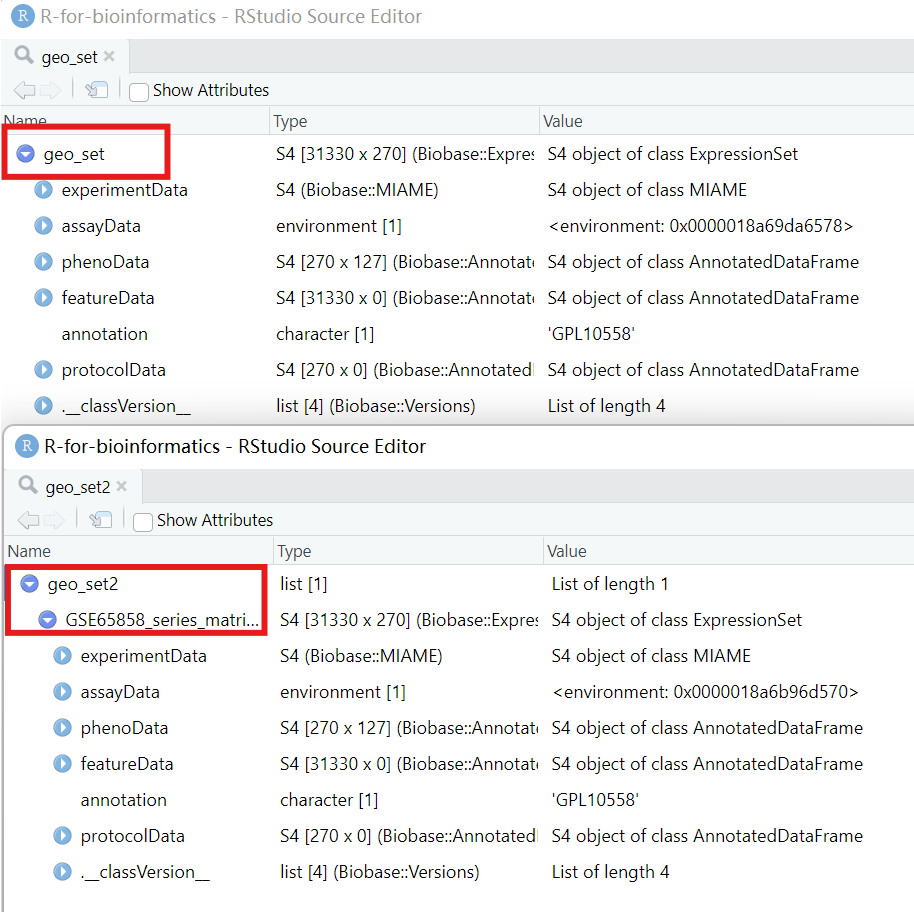
对于第一种(本地下载数据集)应使用:
expr <- exprs(geo_set);
pd <- pData(geo_set);
TCGA数据库
是一个关于癌症研究的数据库
第一种下载方法:从TCGA官网下载
进入TCGA官网
点击顶部四个标签中的Cohort Builder,可以看到有6个选项框:
-
Program数据来源 -
Project所属项目(项目名称一般指癌症种类) -
Disease Type疾病类型 -
Primary Diagnosis初步诊断 -
Primary Site原发部位 -
Tissue or Organ of Origin发病组织或器官
比如我们想搜索TCGA数据库中关于肝癌的数据,就在Program中选择TCGA,在Project中选择TCGA LIHC(肝癌简称)
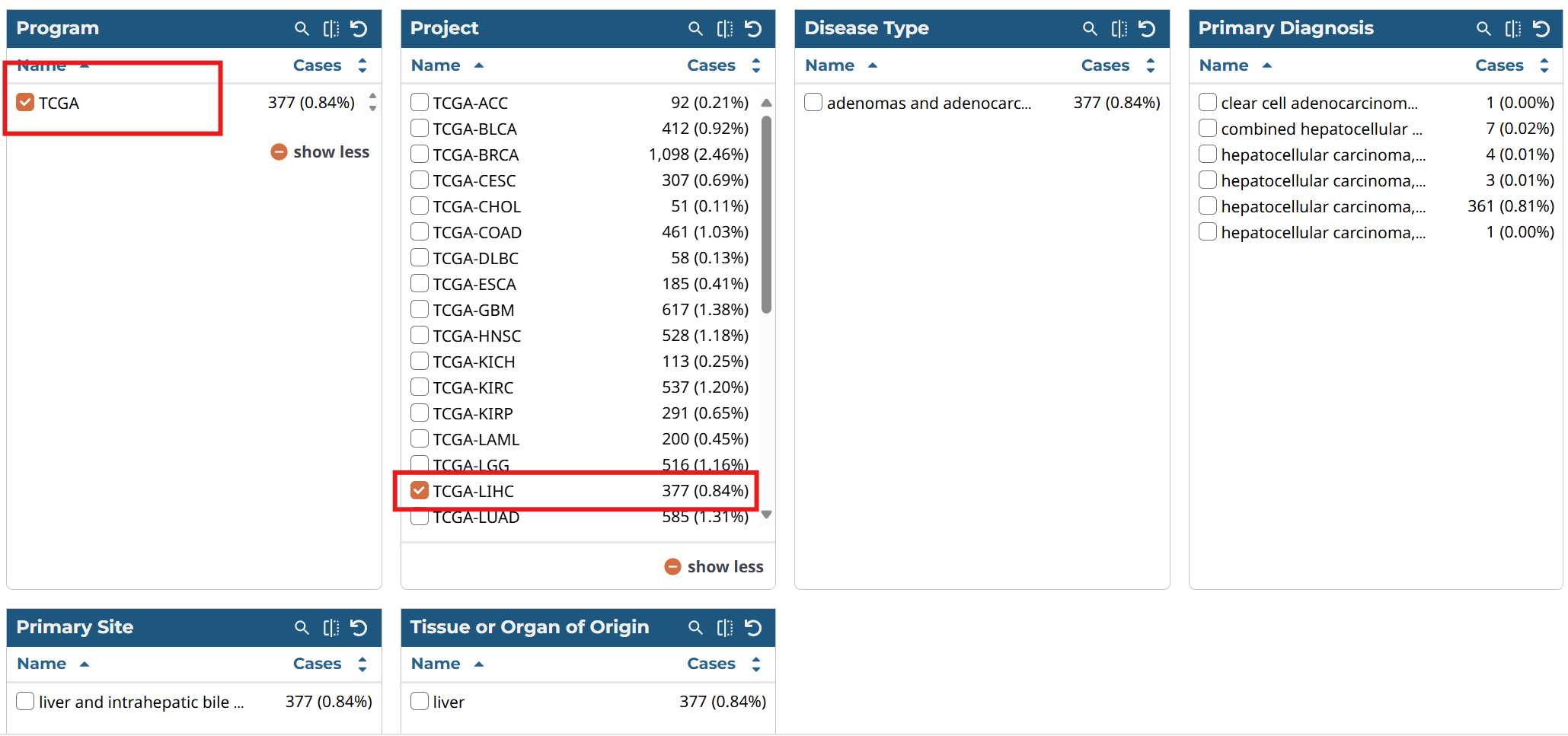
再点击顶部四个标签中的Repository,可以看到左侧边栏中也有一些选项框:
-
Experimental Strategy实验类型 -
Wgs Coverage全基因组测序深度(一般不指定) -
Data Category数据分类 -
Data Type数据类型 -
Data Format数据格式 -
Workflow Type工作流 -
Platform测序平台 -
Access使用权限:controlled不开放,open开放
在Experimental Strategy选择RNA seq,Data Category选择transcriptome profiling,Access选择open
之后点击Add All Files to Cart加数据添加到仓库,再点击右上角的cart即可查看仓库
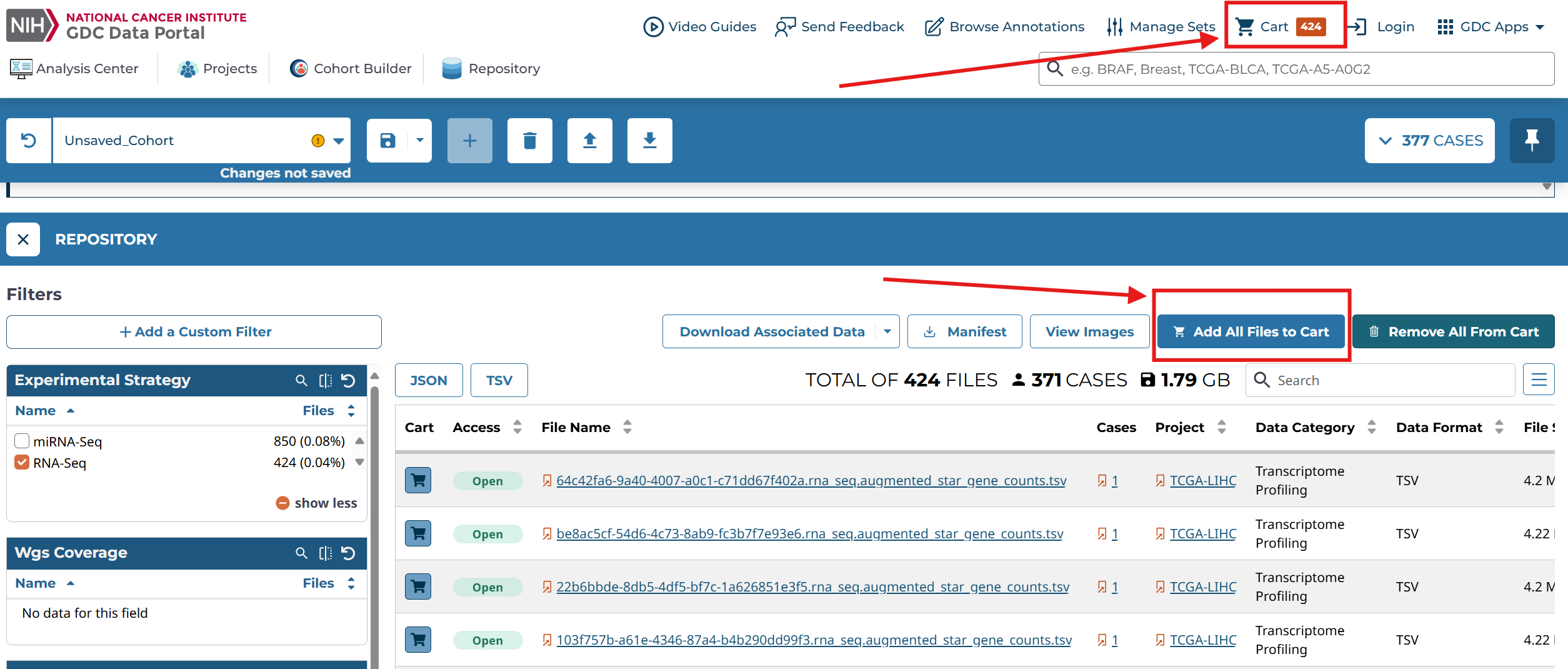
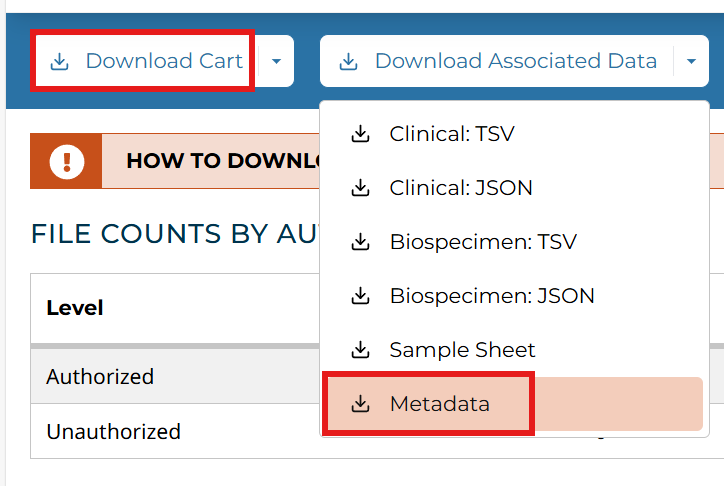
一般情况下要下载两个数据,一个是刚才已添加的cart,另一个是Metadata
第二种下载方法:https://xenabrowser.net/
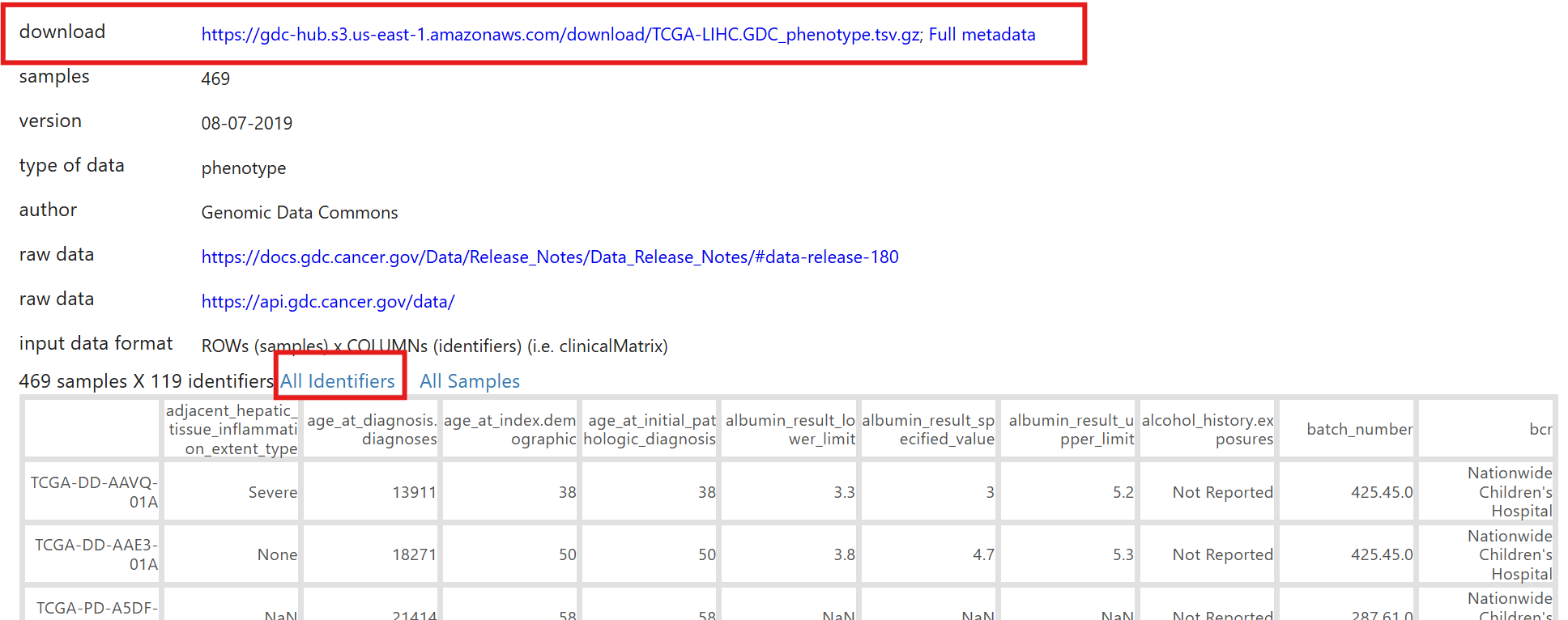
点击顶部的DATA SETS,找到GDC TCGA Liver Cancer (LIHC),点击进入
可以看到有关于它的很多信息,在常规生信中,最关注的就是基因的表达gene expression RNAseq以及临床数据phenotype
点击对应的标签,即可查看详细说明和下载,最下面的表格就是部分数据的展示,点击All Identifiers可以查看所有列
有时从GEO下载的数据会不完整,比如数据作者在他的文章中画了生存曲线,但数据中却没有生存相关的信息。而该网站中的TCGA数据一般较完整
实战
使用数据为TCGA中关于头颈部鳞状细胞癌的counts、临床数据、生存信息
数据预处理
读取及转换成原始值
读取counts数据:
library(data.table);
counts1 <- fread("data\\TCGA-HNSC.htseq_counts.tsv.gz"); # 读取数据
# 因为是.gz格式,所以用fread函数,正常就是read_tsv
counts1:
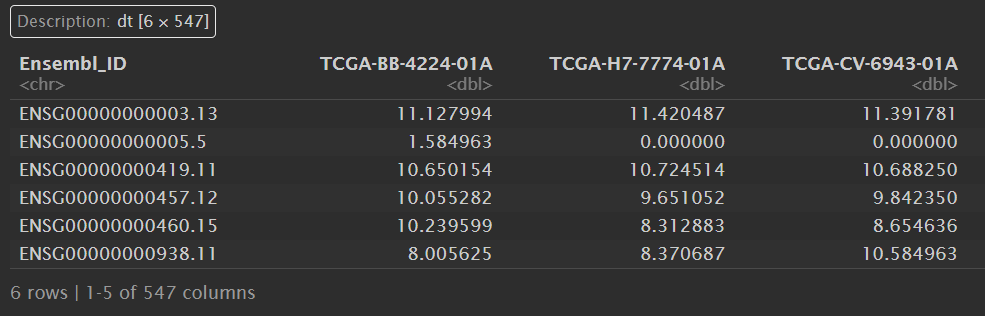
从官网上可知,这里的counts是log2处理后的,一般情况下需要转成原始值再分析
library(tidyverse)
counts1 <- column_to_rownames(counts1, "Ensembl_ID") # 将各基因id转为列名,其它列作为行名
counts1:
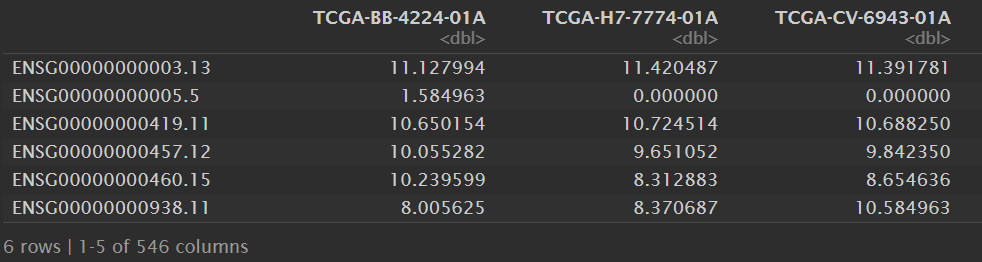
现在要把处理后的counts进行2n操作:
counts2 <- 2 ^ counts1 - 1;
#因为counts无小数,为防止逆转过程中精度损失,需要进行取整操作
counts <- round(counts2);
counts:
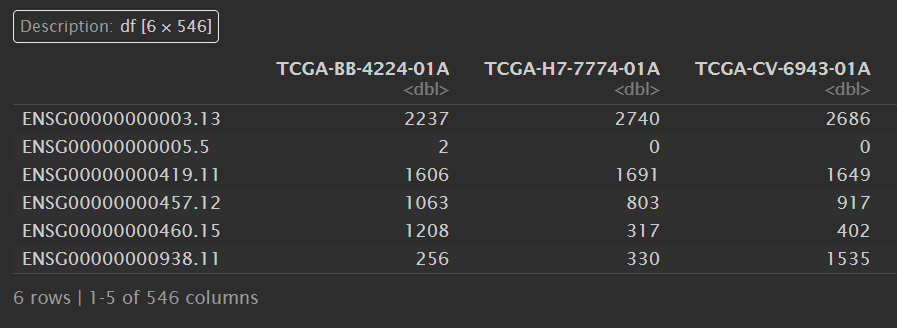
为什么要2n后再-1?
因为从数据库下载下来的counts值,是通过log2对数转换了的,转换之前的原始counts值,有很多值为0,而以2为底,对0进行对数转换的话,是无意义的,数学上也无法转换,所以通常我们在进行log2转换之前,需要先将原始的counts值+1,然后进行对数转换
在counts中,行名为基因id,列名为样本名
读取生存数据和临床数据:
survival_data <- read_tsv("data\\TCGA-HNSC.survival.tsv"); # 生存数据
clinical_data <- fread("data\\TCGA-HNSC.GDC_phenotype.tsv.gz"); # 临床数据
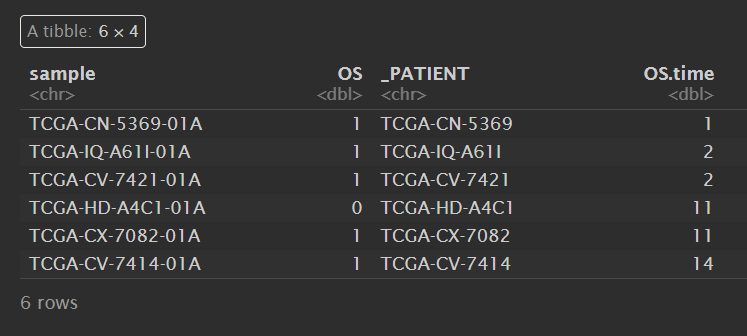
survival_data:
clinical_data:
生存数据中有样本名sample、阳性事件os等列
过滤
过滤掉没有生存数据或者没有表达数据的样本,即对counts和survival_data的样本名取交集
expr_sample <- colnames(counts); # 表达数据的行名(样本名)
surv_sample <- survival_data$sample; # 生存数据的样本名
valid_sample <- intersect(expr_sample, surv_sample); # 取交集,得到既有生存数据也有表达数据的样本
counts <- counts[, valid_sample]; # 取出这些样本(根据列名)
survival_data <- column_to_rownames(survival_data, "sample"); # 样本名的列变行名,方便取出样本
surv_data <- survival_data[valid_sample, ]; # 取出这些样本(根据行名)
counts:
surv_data:
现在我们的表达数据是counts,有545个数据;生存数据是surv_data,也有545个数据
过滤掉非肿瘤样本:
由此需要让样本编号最后三位是01-09的保留
sample_type <- str_split(colnames(counts), pattern = '-', n = 4, simplify = TRUE);
# pattern指定分隔符,n指定分隔成几部分,simplify指定是否将分隔结果组合成向量级
sample_type <- as.data.frame(sample_type); # 转成df
sample_type:
sample_type <- sample_type[, 'V4']; # 只取最后的V4列
unique(sample_type); # "01A" "11A" "01B" "06A"
这说明我们的样本中有”01A”、”11A”、”01B”、”06A”这4中类型
为直观展示是不是肿瘤组织,我们想让”11A”->”normal”,另三种->”tumor”
sample_type <- ifelse(sample_type == "11A", "normal", "tumor");
sample_type <- as.data.frame(sample_type);
sample_type:
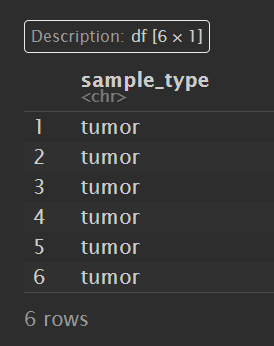
因为counts和surv_data都是直接根据valid_sample取的,所以它们的数据顺序相同,而sample_type是根据counts取的,所以它可以直接应用在surv_data上
is_tumor <- sample_type[, 1] == "tumor";
is_tumor是由bool值组成的vector,第n个元素说明counts中第n列(即surv_data中第n行是不是tumor数据)
tumor_counts <- counts[, is_tumor]; # 根据bool值取列
tumor_surv <- surv_data[is_tumor, ]; # 根据bool值取行
tumor_counts:
tumor_surv:
基因id转基因名
为方便查看数据,将所有的基因id转化成基因名
获取基因id:
gene_id <- rownames(tumor_counts);
gene_id:
因为基因id中.后的没有用,所以删去,方法是先按.分成两部分,再取第一部分
gene_id <- str_split(gene_id, pattern = '[.]', simplify = TRUE); # 为避免'.'被识别成正则表达式,使用'[.]'
gene_id <- gene_id[, 1] # 只取第一列('.'前的部分)
分割后的gene_id:
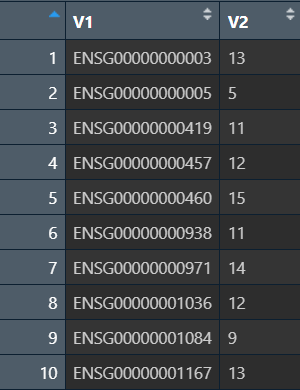
最终结果:
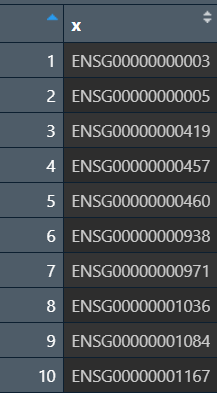
现在我们得到了标准的基因id,需要构建一个tb,一列是基因id,另一列是其对应的基因名
使用org.Hs.eg.db包:
if(!require("org.Hs.eg.db", quietly = T))
{
library("BiocManager");
BiocManager::install("org.Hs.eg.db");
library("org.Hs.eg.db");
}
gene_name <- mapIds(org.Hs.eg.db, gene_id, "SYMBOL", "ENSEMBL");
gene_name:
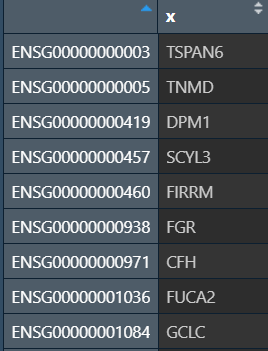
现在基因id是行名,需要变成列名
gene_name <- as.data.frame(gene_name);
gene_name <- rownames_to_column(gene_name, "gene_id");
gene_name:
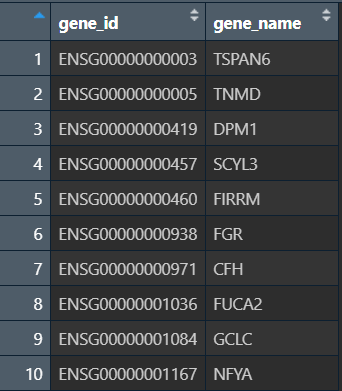
接下来要将tumor_counts中的行名(基因id)变成基因名称
# rownames(tumor_counts) <- gene_name; # 更改counts表的行名(基因id)
# 执行上行代码时报错
为什么会报错:
因为多个基因id可能对应着相同的基因名称,而在counts表中,这些基因id的表达量不同,在转化为基因名称时,为了不让同一个基因名称有多个表达量,需要进行合并操作
另一种方式:不仅可以更改counts表的行名,还能同时让相同的基因对应的表达量合并(取平均值)。
# 先将counts表行名变列名
tumor_counts <- rownames_to_column(tumor_counts, "gene_id"); # 所有的行名(基因id)变为新列gene_id
tumor_counts$gene_id <- str_split(tumor_counts$gene_id, pattern = '[.]', simplify = TRUE)[, 1]; # 将基因id中`.`后的部分去掉,使其格式同gene_name中的基因id
tumor_counts:
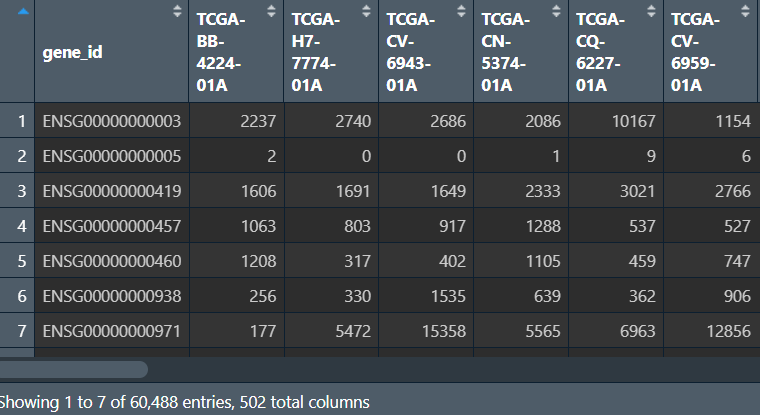
现在观察到tumor_counts和gene_name都有gene_id这列,可以将它们进行连接(join函数)
tumor_counts <- left_join(tumor_counts, gene_name, by = "gene_id"); # 也可以用right_join等
tumor_counts <- relocate(tumor_counts, gene_name, .after = gene_id); # 为方便查看,现将基因名称列调到gene_id列后
tumor_counts:
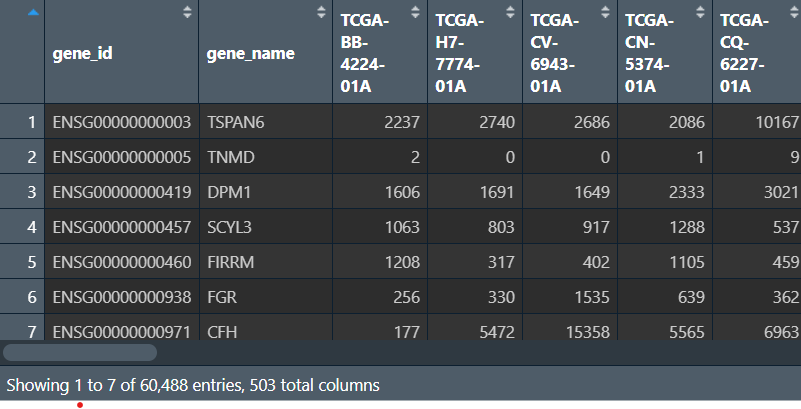
最关键的一步:将同一个基因名称的多个表达量取平均值
注:因为数据量很大,这段代码执行时间可能需要十分钟左右
# 先将基因id转成行名
tumor_counts <- column_to_rownames(tumor_counts, "gene_id");
# 按相同gene_name分组,对每组取平均值
tumor_counts_agg <- aggregate(.~gene_name, FUN = mean, data = tumor_counts);
tumor_counts_agg:
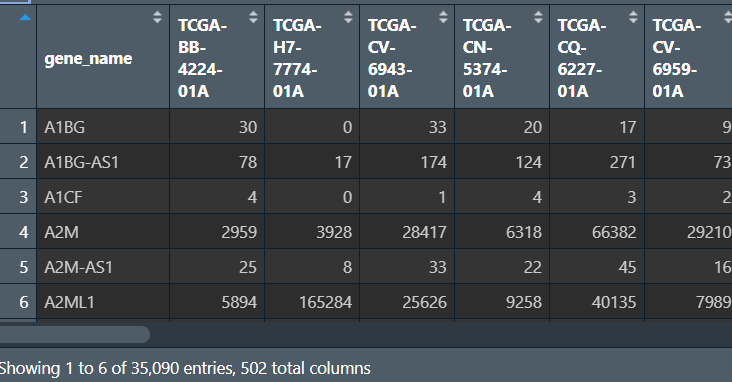
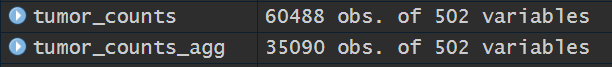
可以看到tumor_counts_agg比tumor_counts少了很多行(60488->35090),且原行名基因id就被删除了
最后将基因名转成行名:
tumor_counts_agg <- column_to_rownames(tumor_counts_agg, "gene_name");
tumor_counts_agg:
代码汇总
为下节课准备:
-
完成初步筛选(既有生存数据也有表达数据的)、未进行肿瘤样本筛选的
surv_data生存数据 -
完成初步筛选、未进行肿瘤样本筛选的、完成基因id转基因名的、取平均值后的
counts_agg数据 -
标明是哪种组织的sample_type
rm(list=ls());
library(data.table);
counts1 <- fread("data\\TCGA-HNSC.htseq_counts.tsv.gz"); # 替换成自己的下载路径
library(tidyverse)
counts1 <- column_to_rownames(counts1, "Ensembl_ID")
counts2 <- 2 ^ counts1 - 1;
counts <- round(counts2);
survival_data <- read_tsv("data\\TCGA-HNSC.survival.tsv"); # 替换成自己的下载路径
expr_sample <- colnames(counts);
surv_sample <- survival_data$sample;
valid_sample <- intersect(expr_sample, surv_sample);
counts <- counts[, valid_sample];
survival_data <- column_to_rownames(survival_data, "sample");
surv_data <- survival_data[valid_sample, ];
sample_type <- str_split(colnames(counts), pattern = '-', n = 4, simplify = TRUE);
sample_type <- as.data.frame(sample_type);
sample_type <- sample_type[, 'V4'];
sample_type <- ifelse(sample_type == "01A", "tumor",
ifelse(sample_type == '11A', 'normal', 'other'));
sample_type <- as.data.frame(sample_type);
gene_id <- rownames(counts);
gene_id <- str_split(gene_id, pattern = '[.]', simplify = TRUE);
gene_id <- gene_id[, 1]
if(!require("org.Hs.eg.db", quietly = T))
{
library("BiocManager");
BiocManager::install("org.Hs.eg.db");
library("org.Hs.eg.db");
}
gene_name <- mapIds(org.Hs.eg.db, gene_id, "SYMBOL", "ENSEMBL");
gene_name <- cbind(gene_name);
gene_name <- as.data.frame(gene_name);
gene_name <- rownames_to_column(gene_name, "gene_id");
counts <- rownames_to_column(counts, "gene_id");
counts$gene_id <- str_split(counts$gene_id, pattern = '[.]', simplify = TRUE)[, 1];
counts <- left_join(counts, gene_name, by = "gene_id");
counts <- relocate(counts, gene_name, .after = gene_id);
counts <- column_to_rownames(counts, "gene_id");
counts_agg <- aggregate(.~gene_name, FUN = mean, data = counts);
counts_agg <- column_to_rownames(counts_agg, "gene_name");
# 存储数据
save(counts_agg, file = "data\\counts.rda");
save(sample_type, file = "data\\sample_type.rda");
save(surv_data, file = "data\\surv_data.rda");
差异表达分析
数据读取及分组
rm(list=ls());
load("data\\counts.rda");
load("data\\surv_data.rda");
load("data\\sample_type.rda");
if(exists("counts_agg")) # 为方便展示,用counts代替counts_agg
{
counts <- counts_agg;
rm(counts_agg)
}
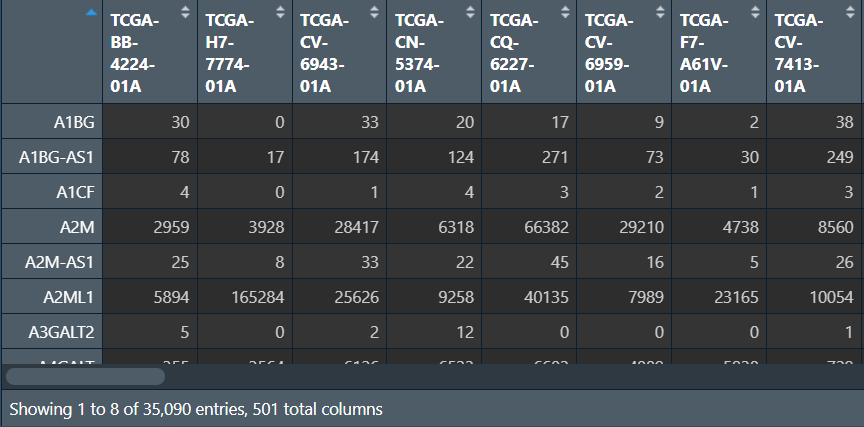
counts:行名是基因名,列名是样本id,每个数据是基因在样本的表达量。我们在上节中取的平均值是对同基因名的行取平均值,使同一基因名只对应一组表达量
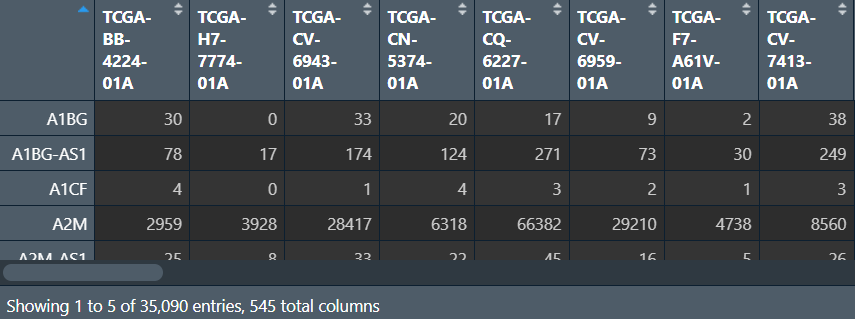
surv_data:行名是样本id,列名是os等生存信息
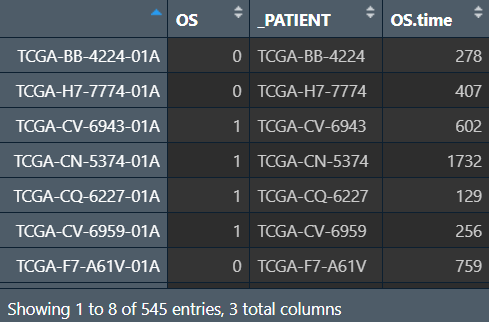
sample_type:一个df,用来标识每个样本的类型,与counts的列名对应,即sample_type的第n个数据表明counts的第n列和surv_data第n行的类型
现在我们要将counts和surv_data分成肿瘤组和正常组:
is_tumor <- sample_type[, 1] == 'tumor'; # 每个样本是不是肿瘤样本
is_normal <- sample_type[, 1] == 'normal'; # 每个样本是不是正常样本
counts_tumor <- counts[, is_tumor]; # 取出所有的肿瘤样本
counts_normal <- counts[, is_normal]; # 取出所有的正常样本
surv_tumor <- surv_data[is_tumor, ];
surv_normal <- surv_data[is_normal, ];
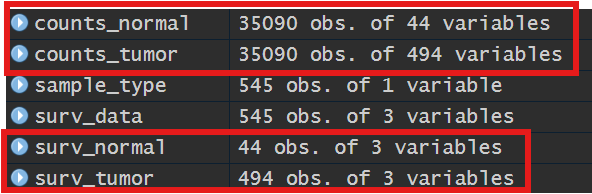
差异分析前的数据分组:
首先将肿瘤组随机分为两等份,一份与正常组织联合起来作差异分析,另一份用于后续的模型验证
使用randomizr包
if(!require("randomizr", quietly = T))
{
library("BiocManager");
BiocManager::install("randomizr");
library("randomizr");
}
grp <- complete_ra(494, num_arms = 2, conditions = c('training', 'validation'));
# 表示将494个样本随机分成两组,分别为training训练组和validation验证组
可以看到得到了一个索引,标志第n个样本被分到了哪组
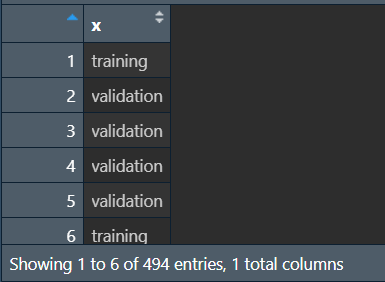
进一步简化成像is_tumor一样的bool值数组:
is_training <- grp == 'training';
is_validation <- grp == 'validation';
将counts_tumor和surv_tumor分为训练和验证组,其中训练组要与正常组(counts_normal和surv_normal)合并作差异分析
counts_tumor_traning <- counts_tumor[, is_training];
# 取出一部分列作为训练组
counts_traning <- cbind(counts_tumor_traning, counts_normal); # 进行合并(注意是增加列),得到训练组
counts_traning <- round(counts_traning); # 取整数,以便后续差异分析(上一节末尾取平均值时可能出现小数)
counts_validation <- counts_tumor[, is_validation];
# 生存信息处理方式同上,区别是筛选行、合并时增加行
# 这里给每个样本都添加一个标记信息,标明是肿瘤组的还是正常组的,方便后续操作
surv_tumor$type <- "tumor";
surv_normal$type <- "normal";
surv_tumor_training <- surv_tumor[is_training, ];
surv_traning <- rbind(surv_tumor_training, surv_normal);
surv_validation <- surv_tumor[is_validation, ];
得到训练组(counts_traning和surv_traning)以及验证组(counts_validation和surv_validation),它们的数据格式相比counts和surv_data不变(只是对counts和surv_data进行取行)
结果中训练组都有291个数据(肿瘤组的训练组247+正常组44),验证组有247个数据,counts和生存信息的都按照grp的方式分组,是肿瘤还是正常、是训练还是验证都相互对应
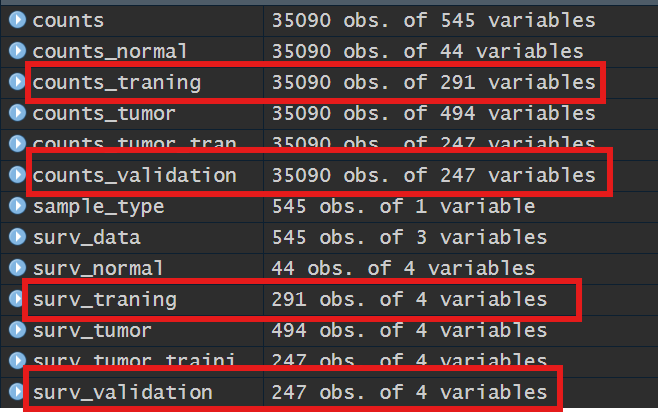
差异分析
使用DESeq2包
if(!require("DESeq2", quietly = T))
{
library("BiocManager");
BiocManager::install("DESeq2");
library("DESeq2");
}
准备分组信息diff_group:
diff_group <- surv_traning[, 'type']; # 使用上面的标记信息,获取训练组每个位置的数据是肿瘤还是正常
diff_group <- as.data.frame(diff_group);
colnames(diff_group) <- c("type"); # 重命名列
diff_group$type <- as.factor(diff_group$type); # 转成factor以适配差异分析函数
diff_group:
进行差异分析:
-
使用
DESeqDataSetFromMatrix等函数拼接要进行差异分析的对象 -
使用
DESeq函数对得到的对象进行差异分析 -
使用
results函数对结果再次汇总分析 -
使用
subset等函数对分析结果进行筛选 -
筛选后结果的列名即为差异基因名
dds <- DESeqDataSetFromMatrix(
countData = counts_traning, # 差异分析的数据
colData = diff_group, # 分组信息
design = ~type # 分组信息中哪列是标志列
); # 获取要分析的对象dds
dds <- DESeq(dds); # 进行分析
res <- results(dds, alpha = 0.05) # 对结果进行分析,设定FDR=0.05
resSig <- subset(res, (abs(log2FoldChange) > 0.58) & padj < 0.05); # 结果筛选
resSig <- as.data.frame(resSig); # 为方便查看转成df格式
diff_gene <- rownames(resSig); # 得到差异表达的基因名
padj设定p值;FoldChange表示基因差异的大小,理论上只要绝对值>1就表示有差异,此时log2FoldChange>0,这里为了筛选掉那些差异化表达很小的基因,所以取>0.58
resSig:
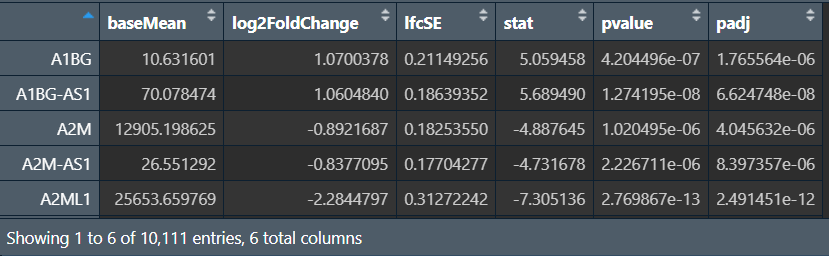
diff_gene:
注:由于分组的随机性,这里得到的resSig和diff_gene每次运行结果可能不同,导致最终筛选出的差异基因数量/名称有少量不同
ADME基因
获取HNSC的ADME基因列表(在资料的Table S1.xlsx中):使用readxl包中的函数读取该文件(其中包含各种癌症的ADME基因)
if(!require("readxl", quietly = T))
{
install.packages("readxl");
library("readxl");
}
adme <- read_excel("data\\Table S1.xlsx", sheet = "ADME genes in 21 cancers");
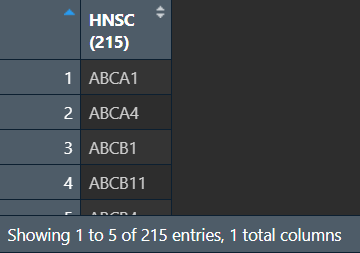
adme_HNSC <- adme[, 7]; # 取到HNSC的adme基因
adme_HNSC <- na.omit(adme_HNSC); # 去除空值
adme_HNSC <- as.matrix(adme_HNSC); # 转换成matrix,方便取交集(只有一列的matrix相当于数组)
adme_HNSC:
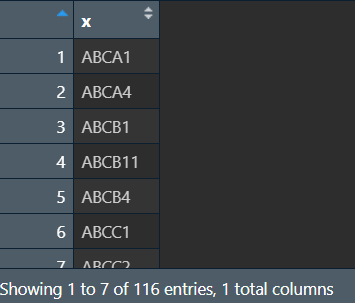
获取差异化表达的adme基因,即对ADME基因与差异化表达基因取交集
special_gene <- intersect(adme_HNSC, diff_gene);
special_gene:
代码汇总
为之后的课程准备:
-
得到的差异基因
special_gene -
差异分析结果
resSig、dds -
肿瘤组的训练组
counts_tumor_traning
rm(list=ls());
load("data\\counts.rda");
load("data\\surv_data.rda");
load("data\\sample_type.rda");
if(exists("counts_agg"))
{
counts <- counts_agg;
rm(counts_agg)
}
is_tumor <- sample_type[, 1] == 'tumor';
is_normal <- sample_type[, 1] == 'normal';
counts_tumor <- counts[, is_tumor];
counts_normal <- counts[, is_normal];
surv_tumor <- surv_data[is_tumor, ];
surv_normal <- surv_data[is_normal, ];
if(!require("randomizr", quietly = T))
{
library("BiocManager");
BiocManager::install("randomizr");
library("randomizr");
}
grp <- complete_ra(494, num_arms = 2, conditions = c('training', 'validation'));
is_training <- grp == 'training';
is_validation <- grp == 'validation';
counts_tumor_traning <- counts_tumor[, is_training];
counts_traning <- cbind(counts_tumor_traning, counts_normal);
counts_traning <- round(counts_traning);
counts_validation <- counts_tumor[, is_validation];
surv_tumor$type <- "tumor";
surv_normal$type <- "normal";
surv_tumor_training <- surv_tumor[is_training, ];
surv_traning <- rbind(surv_tumor_training, surv_normal);
surv_validation <- surv_tumor[is_validation, ];
if(!require("DESeq2", quietly = T))
{
library("BiocManager");
BiocManager::install("DESeq2");
library("DESeq2");
}
diff_group <- surv_traning[, 'type'];
diff_group <- as.data.frame(diff_group);
colnames(diff_group) <- c("type");
diff_group$type <- as.factor(diff_group$type);
dds <- DESeqDataSetFromMatrix(
countData = counts_traning,
colData = diff_group,
design = ~type
);
dds <- DESeq(dds);
res <- results(dds, alpha = 0.05)
resSig <- subset(res, (abs(log2FoldChange) > 0.58) & padj < 0.05);
resSig <- as.data.frame(resSig);
diff_gene <- rownames(resSig);
if(!require("readxl", quietly = T))
{
install.packages("readxl");
library("readxl");
}
adme <- read_excel("data\\Table S1.xlsx", sheet = "ADME genes in 21 cancers"); # 替换成自己的资料路径
adme_HNSC <- adme[, 7];
adme_HNSC <- na.omit(adme_HNSC);
adme_HNSC <- as.matrix(adme_HNSC);
special_gene <- intersect(adme_HNSC, diff_gene);
# 存储数据
save(special_gene, file = "data\\special_gene.rda");
save(resSig, file = "data\\resSig.rda");
save(dds, file = "data\\dds.rda");
save(counts_tumor_traning, file = "data\\counts_tumor_traning.rda");
GO富集分析
概念介绍
GO富集(Gene Ontology):针对分子功能进行富集,把基因的功能分成了三个部分
-
细胞组分(cellular component,CC)
-
分子功能(molecular function,MF)
-
生物过程(biological process,BP)
简要来说,就是把得到所有基因的所有功能,然后统计功能的种类、以及每种功能有多少种基因
以下为一个GO富集分析结果:
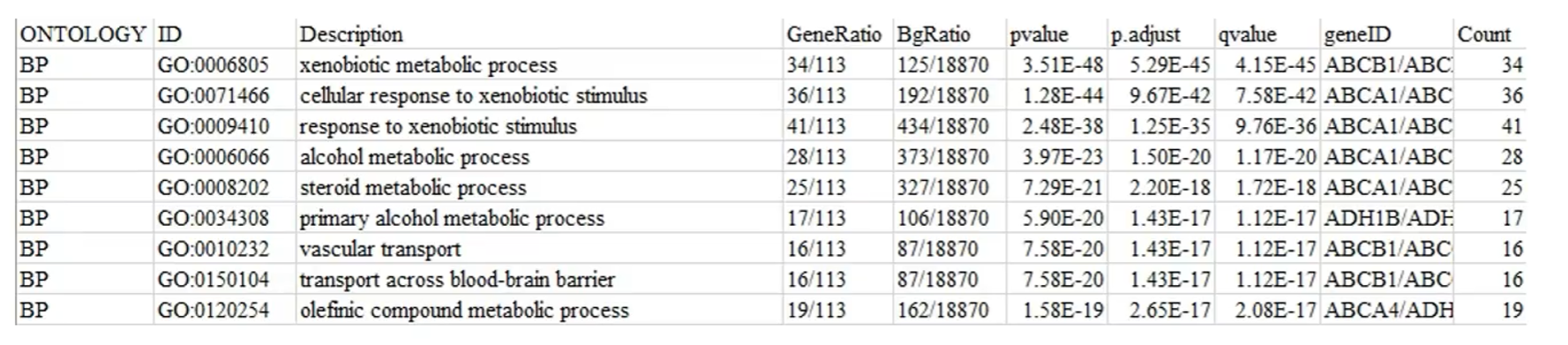
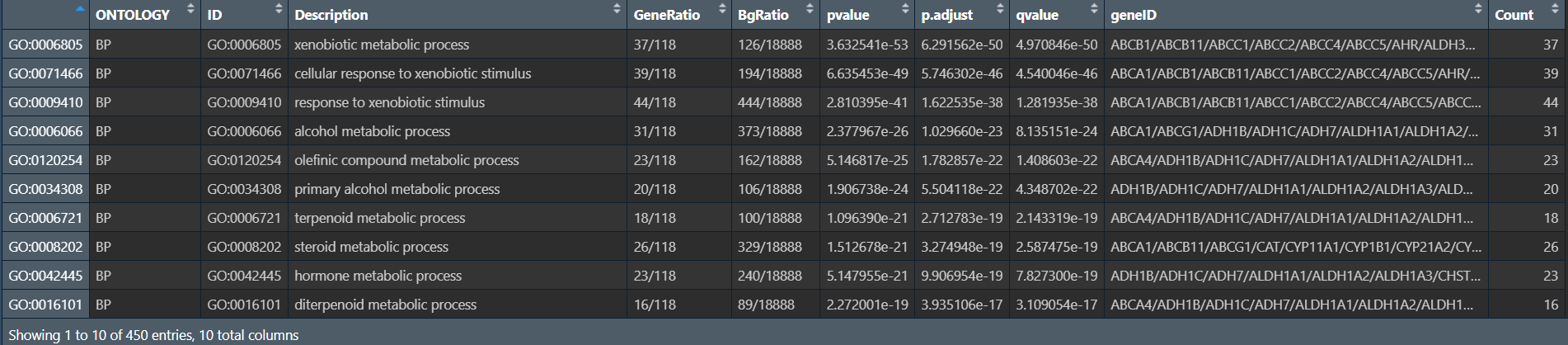
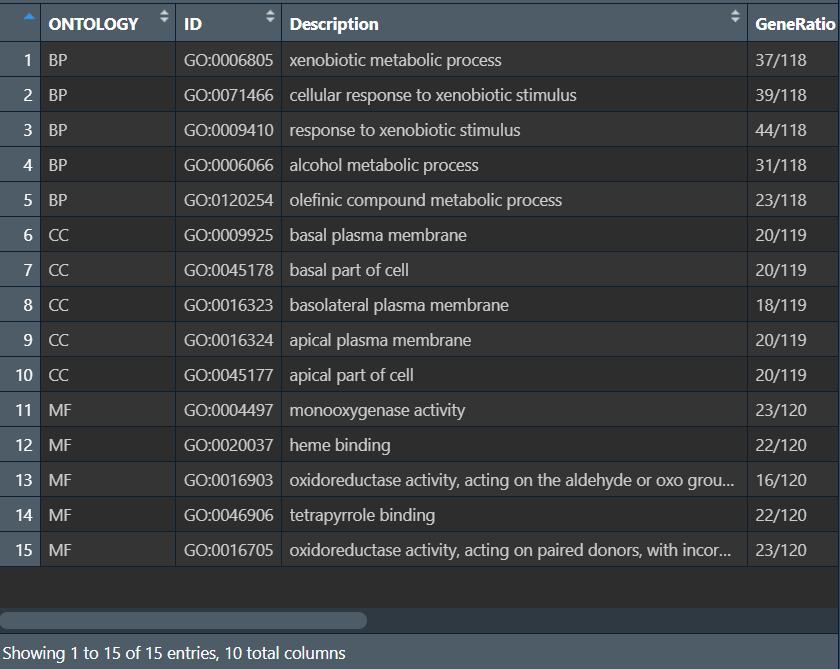
-
ONTOLOGY:即上面提到基因的三个功能 -
ID:具体的GO条目的ID号 -
Description:GO条目的简要描述,比如是哪个细胞组分、哪个分子功能 -
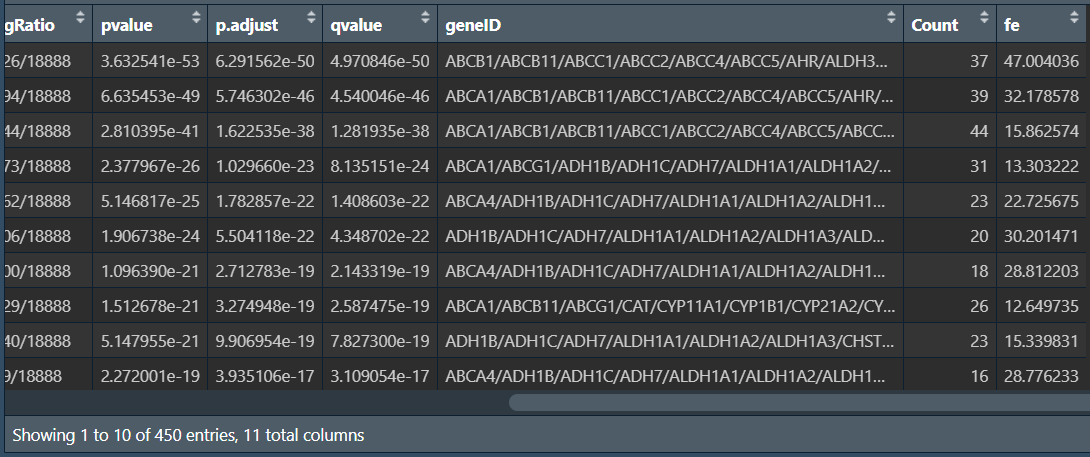
GeneRatio:是一个分数,分子是富集到这个GO条目上基因的数目,分母是所有输入的做富集分析的基因的数目(可以是差异表达分析得到的基因) -
BgRatio(Background Ratio):分母是人所有编码蛋白基因中有GO注释的基因的数目(所有被统计出功能的基因数),分子是这18870个基因中注释到这个GO条目上的基因数目(有该功能的基因数目) -
富集倍数(Fold enrichment):
GeneRatio/BgRatio -
pvalue:富集的p值。P值越小,表明结果越显著/可信 -
p.adjust:校正之后的p值,相对于p-value可信度更高 -
qvalue:q值,可以简单理解为p-value产生假阳性的概率,也是是p值校正后的结果,越小则结果越可信。它比p-value更加严格,可能过滤掉少部分阳性结果 -
geneID:样本基因中富集到这个GO条目上面的具体基因名字 -
count:样本基因中富集到这个GO条目上的基因的数目,即GeneRatio的分子
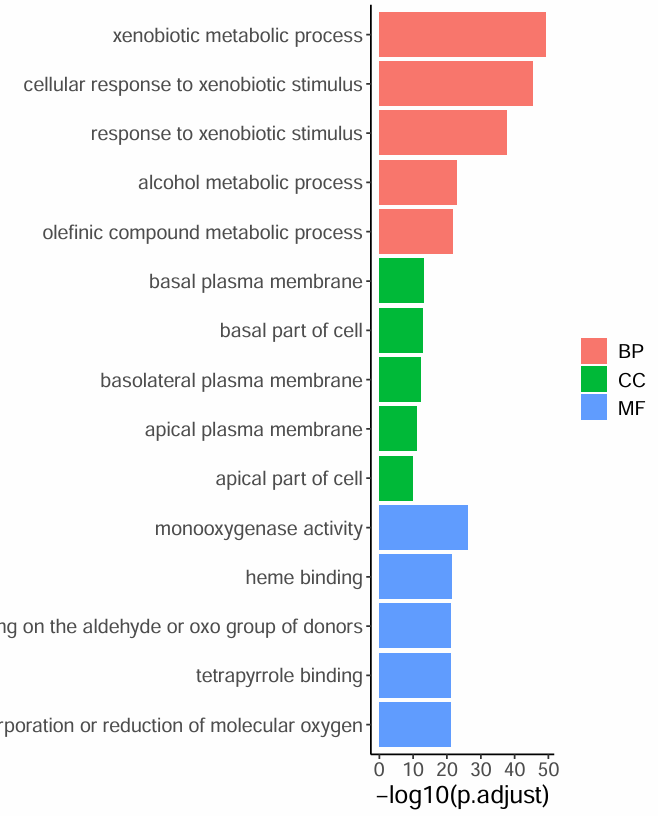
4种可视化图:
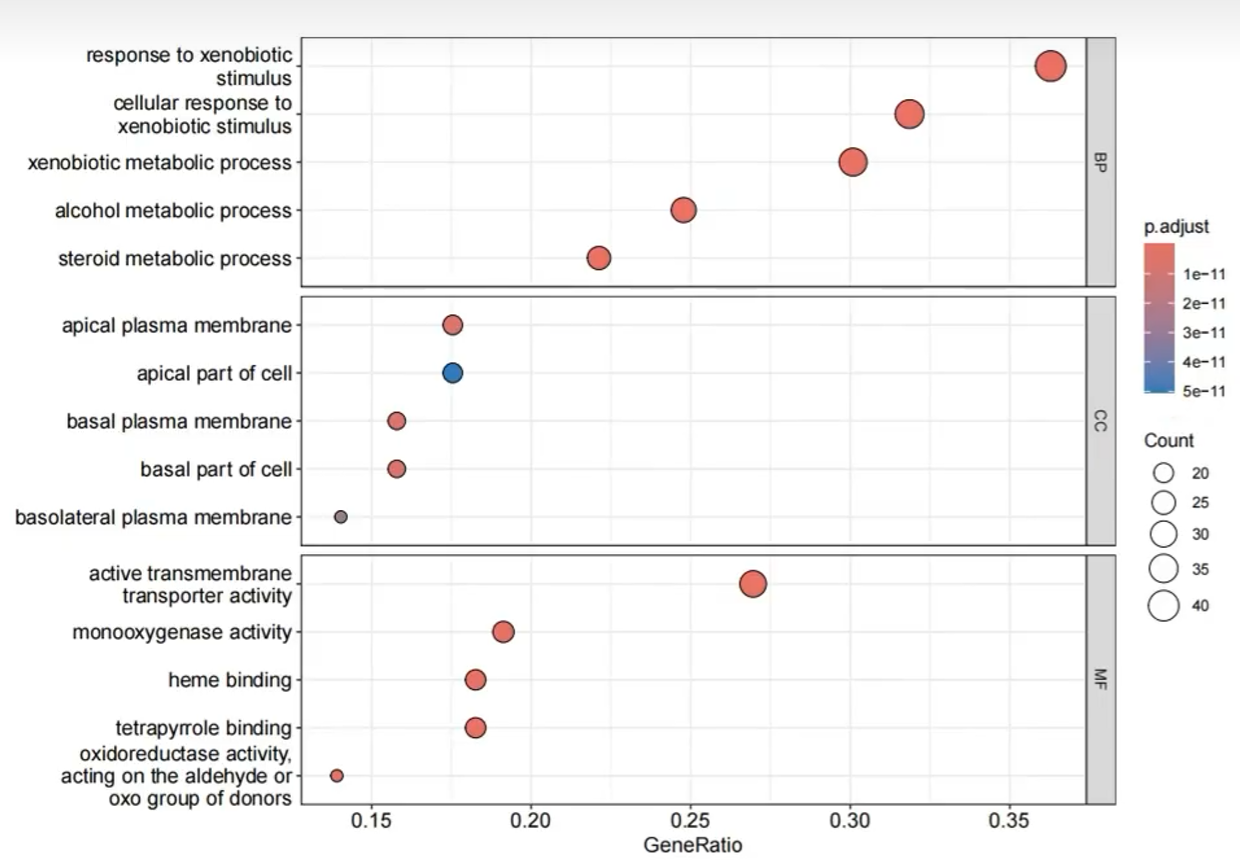
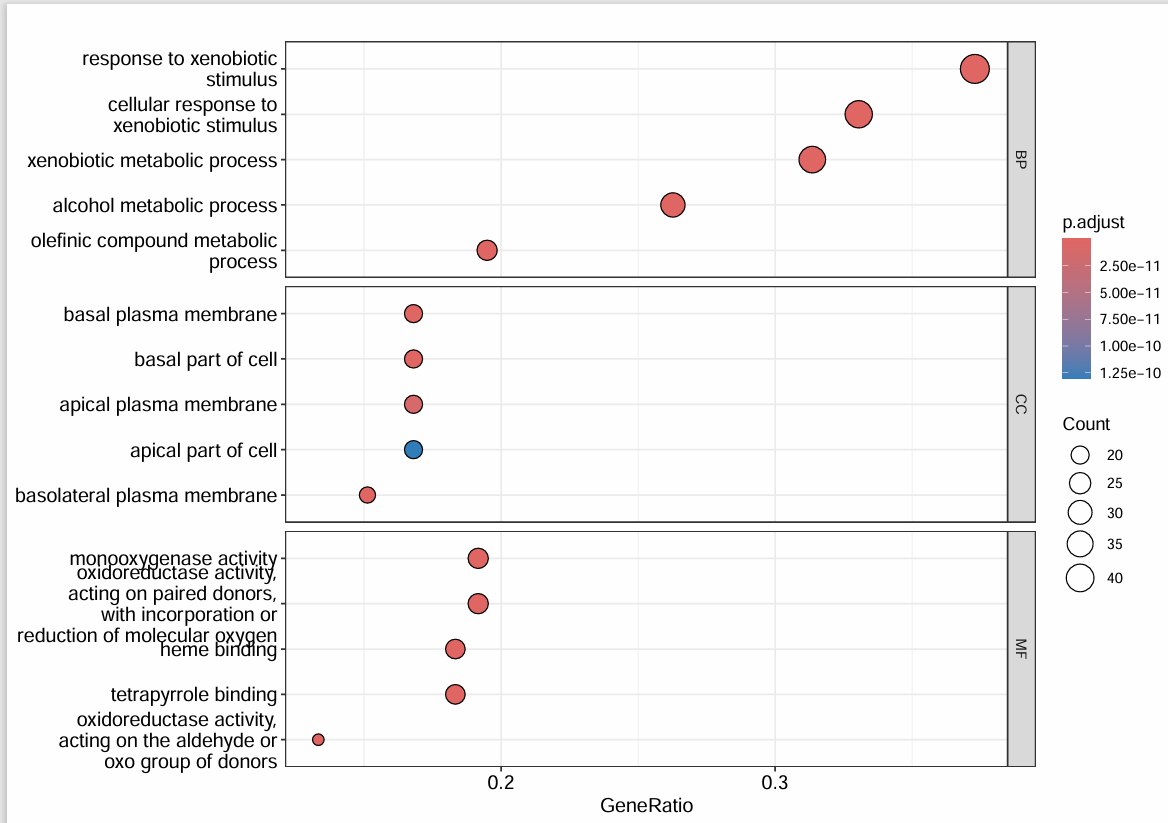
是气泡图,横坐标是GeneRatio,纵坐标是Description,分成了三个框,代表三种基因功能。点越大则有该功能的数量越多,颜色越红则p值越小
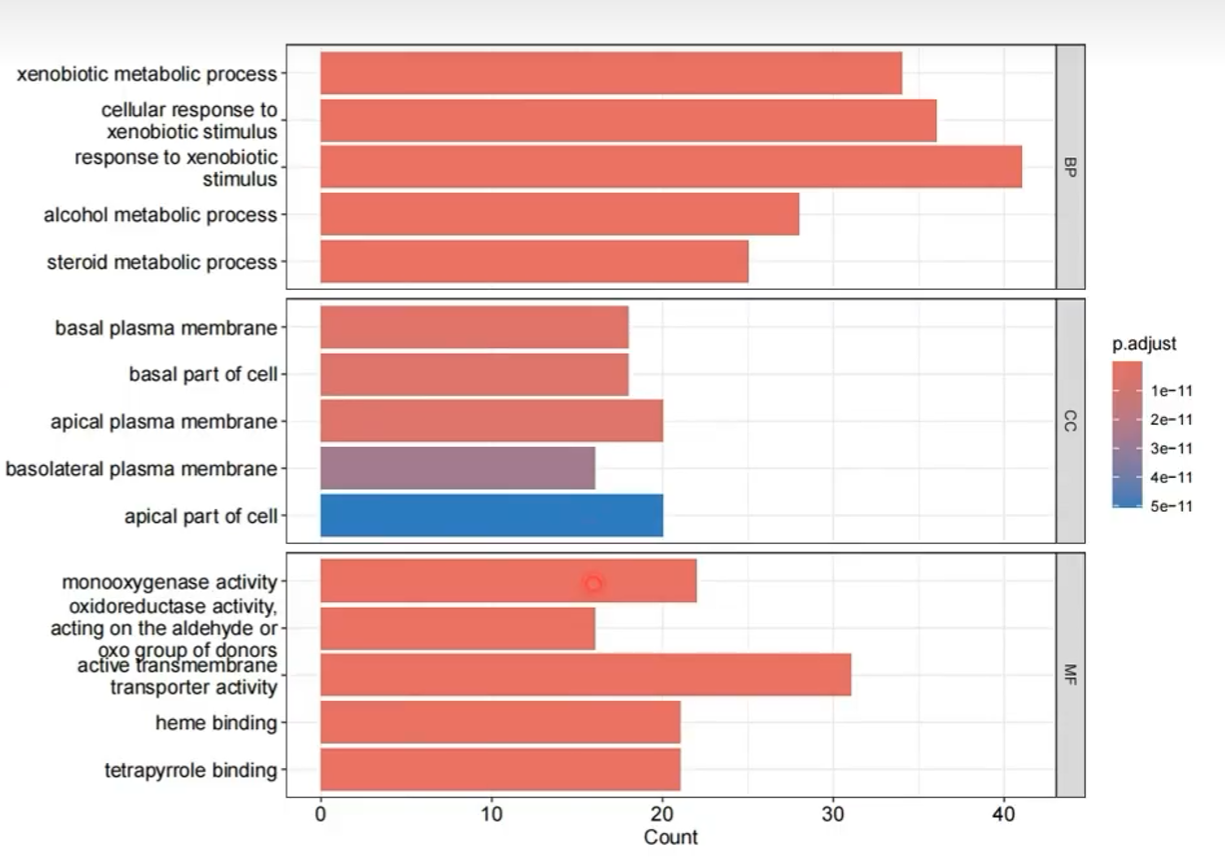
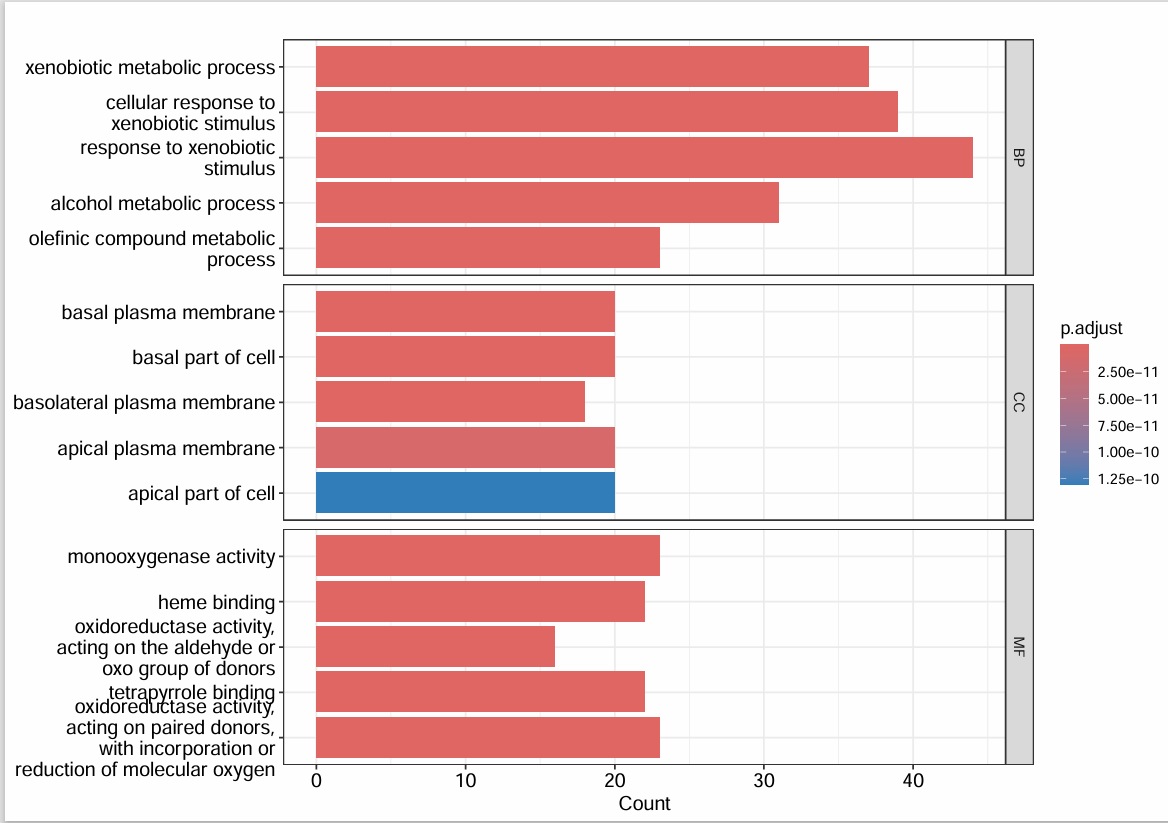
是柱状图,横坐标是count,纵坐标是Description,也分成了三个框,代表三种基因功能。柱状条越大则有该功能的数量越多,颜色越红则p值越小
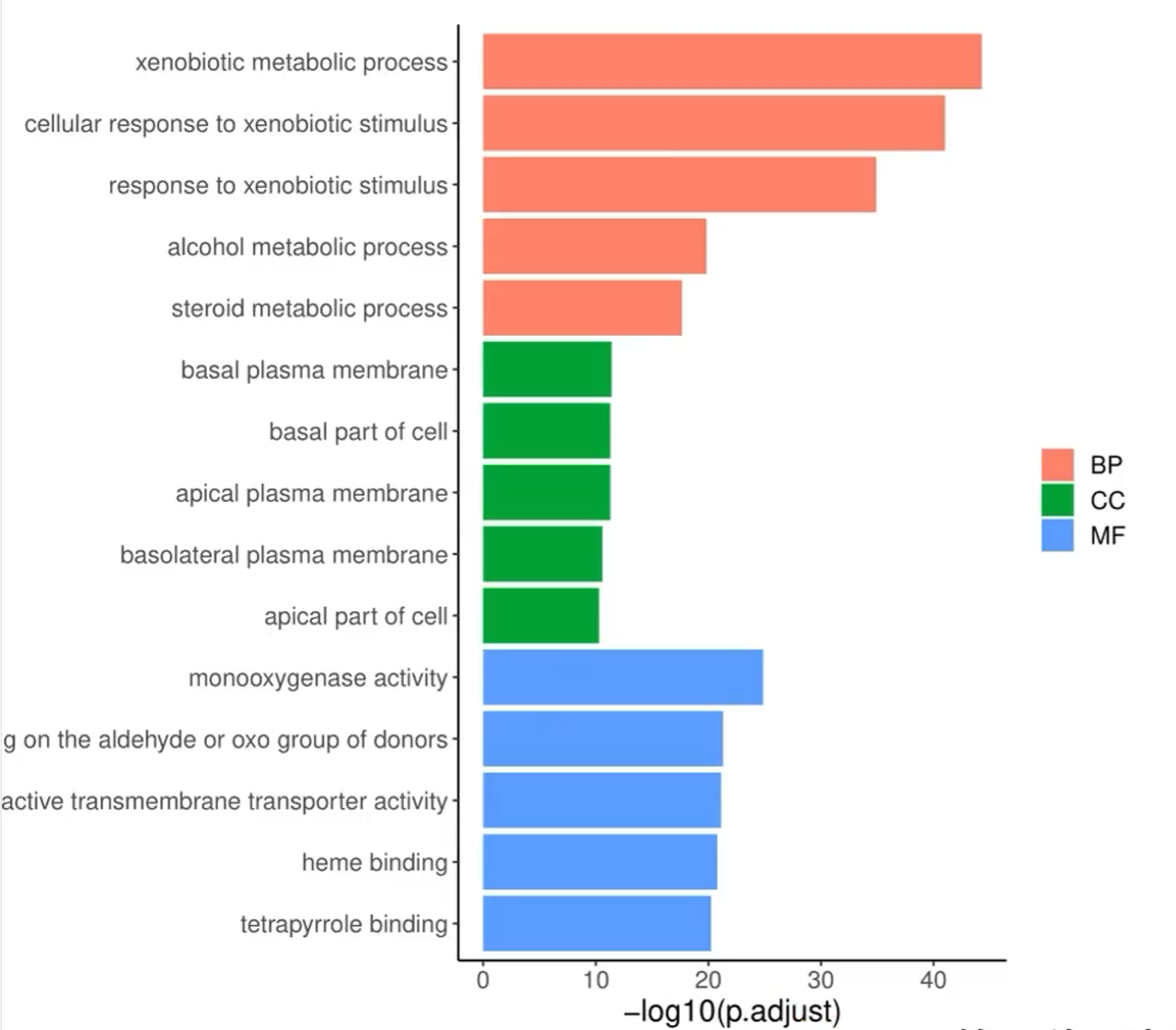
是柱状图,横坐标是p值的负对数,纵坐标是Description,用三种颜色表示三种功能
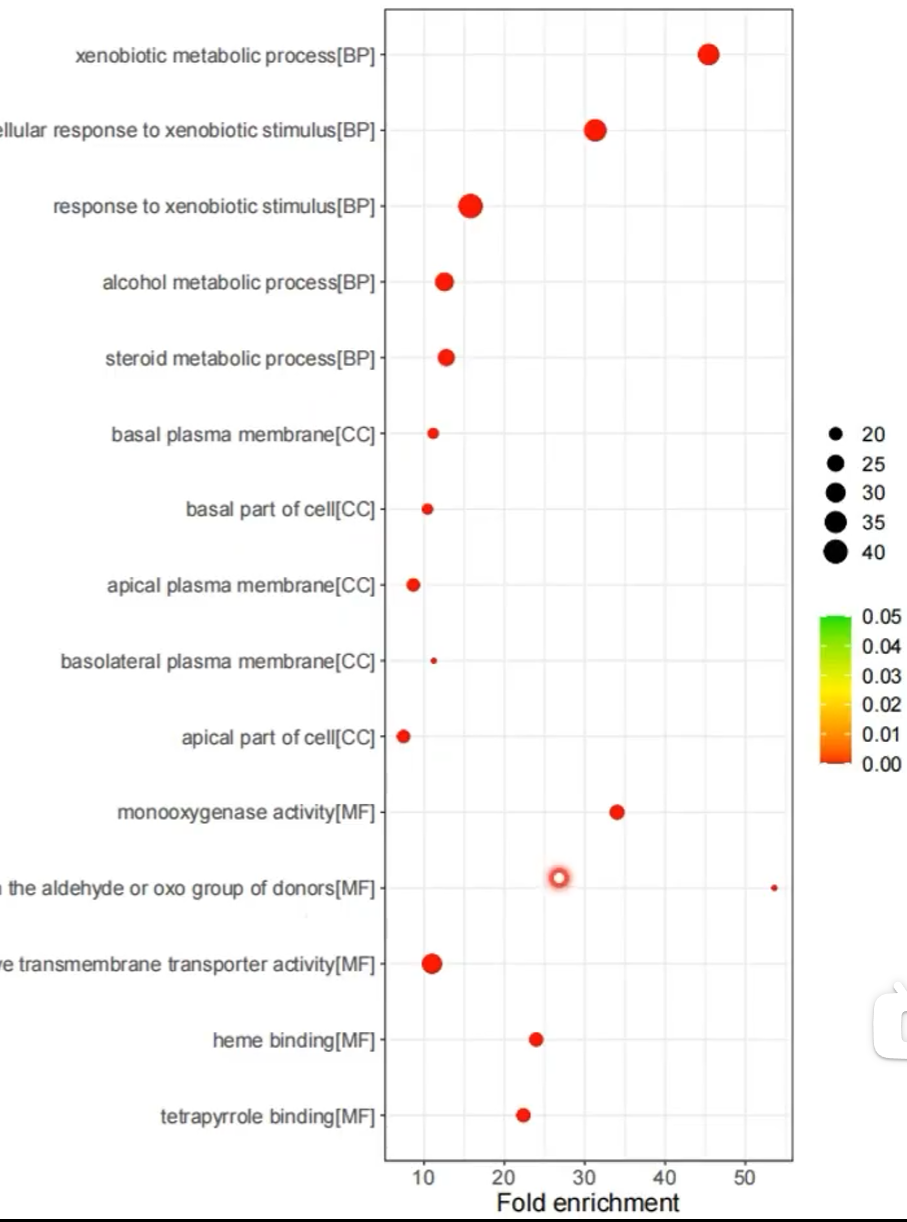
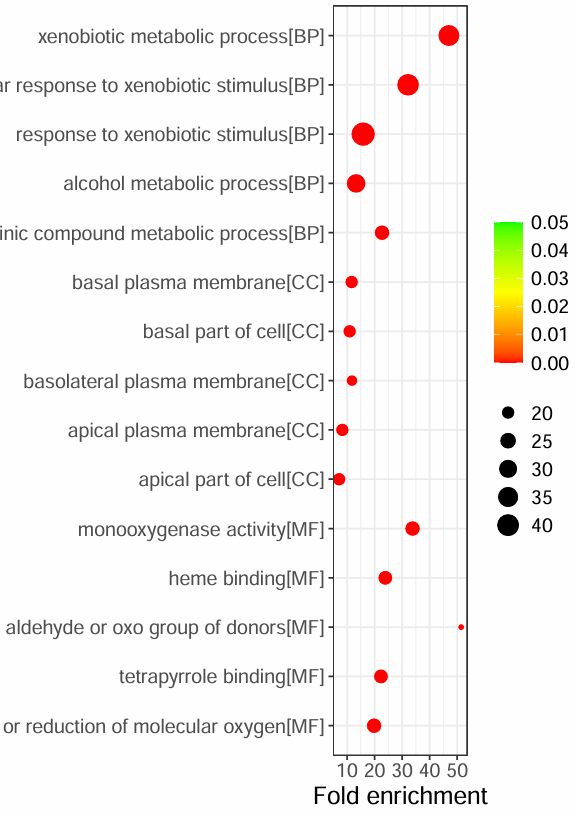
是气泡图,横坐标是Fold enrichment即富集倍数,纵坐标是Description,每种描述后都标注了属于那种基因功能。点越大则有该功能的数量越多,颜色越红则p值越小
进行分析
使用clusterProfiler包
if(!require("clusterProfiler", quietly = T))
{
library("BiocManager");
BiocManager::install("clusterProfiler");
library("clusterProfiler");
}
载入数据:
load("data\\special_gene.rda");
clusterProfiler包的使用:
library("org.Hs.eg.db"); # go分析中要用这个基因数据库
enrich.go <- enrichGO(
gene = special_gene, # 要GO富集分析的基因
OrgDb = org.Hs.eg.db, # 指定物种的基因数据库
keyType = 'SYMBOL', # 指定基因名称类型,有SYMBOL、ENSEMBL、ENTREZID三种类型
ont = 'ALL', # 要富集的基因功能,可选BP、MF、CC、ALL(都分析)
pAdjustMethod = 'BH', # p值校正方法
minGSSize = 10, # 结果中富集的GO条目至少包含10个基因,少于10的GO忽略
pvalueCutoff = 0.05, # 指定p值阈值<0.05
qvalueCutoff = 0.2, # 指定q值阈值<0.2
readable = FALSE # 是否将基因id转为基因名,因为前面已经转完了,这里就不用再转了
);
go <- as.data.frame(enrich.go);
view(go);
# 存储数据
write.table(
go,
'data\\enrich_go.txt',
row.names = FALSE,
quote = FALSE
);
可以得到像上面提到的GO分析结果:
绘图
使用ggplot2包
library("ggplot2");
第一个图:
pdf(file = "data\\go_all_dot.pdf", width = 10); # 将作图结果保存到PDF文件中
dotplot(enrich.go, # 使用enrich.go数据
split = 'ONTOLOGY', # 按照ONTOLOGY来分组
showCategory = 5) + # 每组显示前5条
facet_grid(ONTOLOGY~., scale = "free"); # 用ONTOLOGY来分框
dev.off(); # 关闭PDF文件
第二个图:与1类似,只是把作图函数换成barplot
pdf(file = "data\\go_all_bar.pdf", width = 10);
barplot(enrich.go, # 使用enrich.go数据
split = 'ONTOLOGY', # 按照ONTOLOGY来分组
showCategory = 5) + # 每组显示前5条
facet_grid(ONTOLOGY~., scale = "free"); # 用ONTOLOGY来分框
dev.off();
第三个图:
先将结果按校正后p值升序排列,并按ONTOLOGY分成BP、MF、CC三组,每组取前5个基因
library("dplyr");
top5 <- enrich.go %>%
group_by(ONTOLOGY) %>%
top_n(n = -5, wt = p.adjust); # 降序排列的后5个基因
top5 <- as.data.frame(top5);
view(top5);
开始画图:
pdf(file = "data\\go_barplot.pdf", width = 10);
ggplot(top5,
aes(x = Description, # x轴是描述
y = -log10(p.adjust), # y轴是p值的负对数
fill = ONTOLOGY)) + # 根据ONTOLOGY类别填充颜色
geom_bar(stat = "identity") + # 画柱状图
coord_flip() + # 交换XY轴位置
scale_x_discrete(limits = rev(top5$Description)) + # 设置x轴各条目的顺序
theme_classic() + # 使用经典主题
theme(
text = element_text(size = 15), # 字体尺寸为15
axis.title.y = element_blank(), # y轴标签为空
axis.title.x = element_text(size = 15), # x轴字体大小为15
legend.title = element_blank() # 图例为空
);
dev.off();
第四个图:
首先计算富集倍数,因为enrich.go中得到的都是字符串型的分数,要先转换成数值型再相除
str_to_num <- function(ratio){ # 将字符串型的分数转换成数值型
sapply(ratio, function(x) as.numeric(strsplit(x, '/')[[1]][1]) / as.numeric(strsplit(x, '/')[[1]][2]))
}
go_fe <- enrich.go %>%
mutate(fe = str_to_num(GeneRatio) / str_to_num(BgRatio)); # 将富集倍数保存到fe列上
go_fe_df <- as.data.frame(go_fe);
view(go_fe_df);
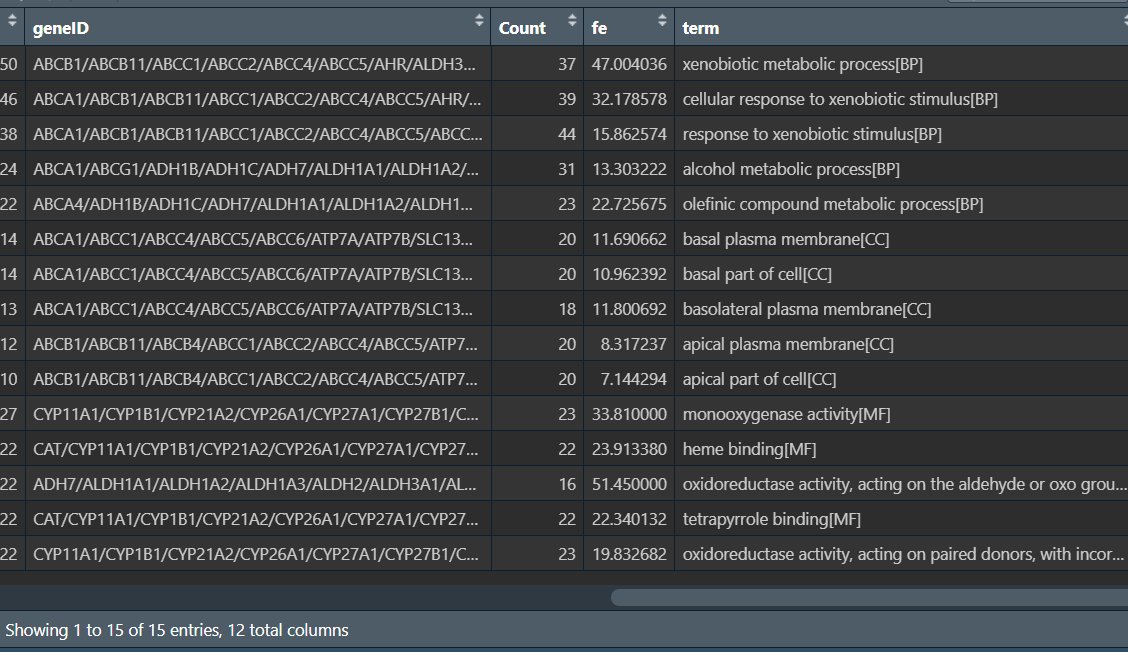
之后与第三个图相同:将结果按校正后p值升序排列,并按ONTOLOGY分成BP、MF、CC三组,每组取前5个基因
# 按照前面相同的方法
top5_fe <- go_fe %>%
group_by(ONTOLOGY) %>%
top_n(n = -5, wt = p.adjust);
最后在每个GO描述后加上对应的BP、MF或CC
top5_fe <- top5_fe %>%
mutate(term = paste0(Description,"[", ONTOLOGY, "]")); # 将新Description保存到term列上
top5_fe <- as.data.frame(top5_fe);
view(top);
开始画图:
pdf(file = "data\\go_dotplot.pdf", width = 10);
ggplot(top5_fe,
aes(x = term, # x轴是新的描述
y = fe, # y轴是富集倍数
color = p.adjust, # 颜色按照p值填充
size = Count)) + # 根据Count数量来确定点的大小
geom_point() + # 画散点图
coord_flip() + # 交换XY轴位置
scale_colour_gradientn(limits = c(0, 0.05), # p值范围
colors = c("red", "yellow", "green")) + # 颜色渐变顺序:p值=0->red,=0.05->green,中间值为yellow
scale_x_discrete(limits = rev(top5_fe$term)) + # 设置x轴各条目的顺序
theme_bw() + # 使用黑白主题
ylab("Fold enrichment") + # y轴标题
theme(
text = element_text(size = 15), # 字体尺寸为15
axis.title.y = element_blank(), # y轴标签为空
axis.title.x = element_text(size = 15), # x轴字体大小为15
legend.title = element_blank() # 图例为空
);
dev.off();
代码汇总
rm(list=ls());
load("data\\special_gene.rda");
if(!require("clusterProfiler", quietly = T))
{
library("BiocManager");
BiocManager::install("clusterProfiler");
library("clusterProfiler");
}
library("org.Hs.eg.db");
enrich.go <- enrichGO(
gene = special_gene,
OrgDb = org.Hs.eg.db,
keyType = 'SYMBOL',
ont = 'ALL',
pAdjustMethod = 'BH',
minGSSize = 10,
pvalueCutoff = 0.05,
qvalueCutoff = 0.2,
readable = FALSE
);
go <- as.data.frame(enrich.go);
# 存储数据
write.table(go, 'data\\enrich_go.txt', row.names = FALSE, quote = FALSE);
library("ggplot2");
# 1
pdf(file = "data\\go_all_dot.pdf", width = 10);
dotplot(enrich.go, split = 'ONTOLOGY', showCategory = 5) +
facet_grid(ONTOLOGY~., scale = "free");
dev.off();
# 2
pdf(file = "data\\go_all_bar.pdf", width = 10);
barplot(enrich.go, split = 'ONTOLOGY', showCategory = 5) +
facet_grid(ONTOLOGY~., scale = "free");
dev.off();
# 3
library("dplyr");
top5 <- enrich.go %>%
group_by(ONTOLOGY) %>%
top_n(n = -5, wt = p.adjust);
top5 <- as.data.frame(top5);
pdf(file = "data\\go_barplot.pdf", width = 10);
ggplot(top5,aes(x = Description,y = -log10(p.adjust),fill = ONTOLOGY)) +
geom_bar(stat = "identity") +
coord_flip() +
scale_x_discrete(limits = rev(top5$Description)) +
theme_classic() +
theme(text = element_text(size = 15),axis.title.y = element_blank(),axis.title.x = element_text(size = 15),legend.title = element_blank());
dev.off();
# 4
str_to_num <- function(ratio){
sapply(ratio, function(x) as.numeric(strsplit(x, '/')[[1]][1]) / as.numeric(strsplit(x, '/')[[1]][2]))
}
go_fe <- enrich.go %>%
mutate(fe = str_to_num(GeneRatio) / str_to_num(BgRatio));
go_fe_df <- as.data.frame(go_fe);
top5_fe <- go_fe %>%
group_by(ONTOLOGY) %>%
top_n(n = -5, wt = p.adjust);
top5_fe <- top5_fe %>%
mutate(term = paste0(Description,"[", ONTOLOGY, "]"));
top5_fe <- as.data.frame(top5_fe);
pdf(file = "data\\go_dotplot.pdf", width = 10);
ggplot(top5_fe,aes(x = term,y = fe,color = p.adjust,size = Count)) +
geom_point() +
coord_flip() +
scale_colour_gradientn(limits = c(0, 0.05),colors = c("red", "yellow", "green")) +
scale_x_discrete(limits = rev(top5_fe$term)) +
theme_bw() +
ylab("Fold enrichment") +
theme(text = element_text(size = 15),axis.title.y = element_blank(),axis.title.x = element_text(size = 15),legend.title = element_blank());
dev.off();
KEGG富集分析
概念介绍
KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库是系统地分析基因功能、链接基因组信息和功能信息的数据库,包括代谢通路(pathway)数据库、分层分类数据库、基因数据库、基因组数据库等
KEGG PATHWAY图片简单介绍:
进入KEGG官网,点击KEGG PATHWAY:
以小细胞肺癌为例,在搜索框内输入small cell lung cancer,
点击前面的id号进入,其中最关注的就是Pathway map
点击图片可以看到放大的图片
图中的箭头表示促进,箭头上如果带有横线就表示抑制
KEGG富集分析的结果可类比GO富集分析
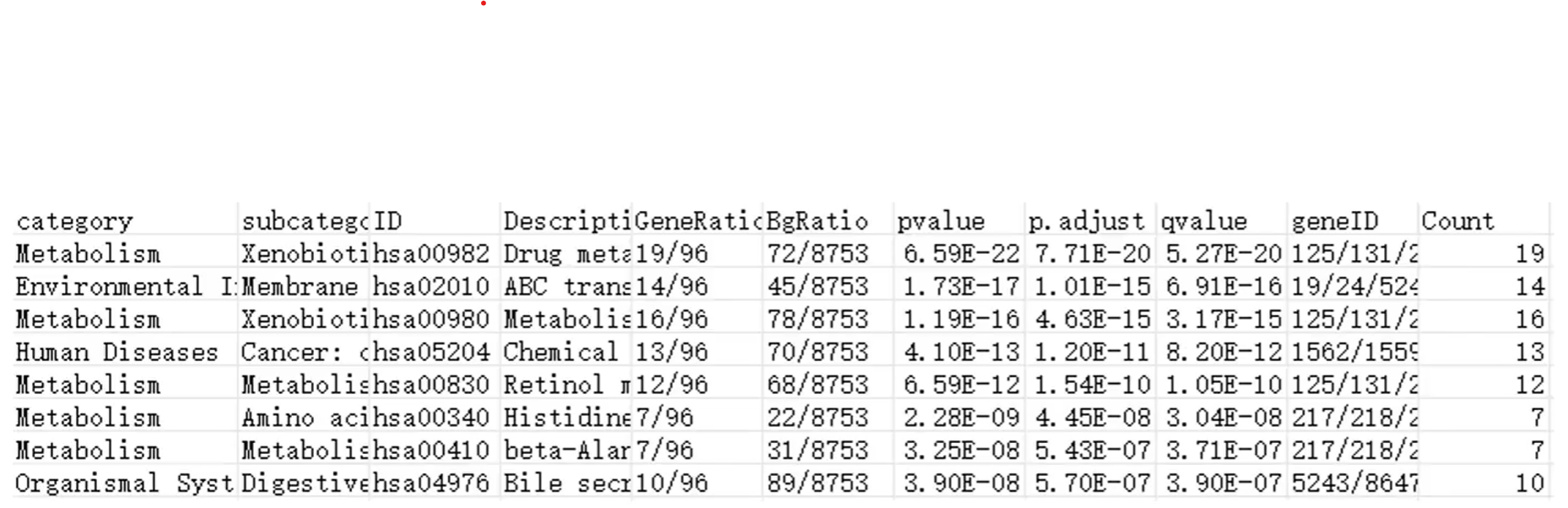
以下为一个KEGG富集分析的结果:
-
category/subcategory:通路的分类/二级分类 -
ID:KEGG通路的ID号 -
Description:KEGG通路的描述 -
GeneRatio:是一个分数,分子是富集到这个KEGG通路上基因的数目,分母是所有输入的做富集分析的基因的数目(可以是差异表达分析得到的基因) -
BgRatio(Background Ratio):分母是人所有基因中有KEGG注释的基因的数目(所有被统计出功能的基因数),分子是这些基因中注释到这个KEGG通路上的基因数目(有该功能的基因数目) -
富集倍数(Fold enrichment):
GeneRatio/BgRatio -
pvalue:富集的p值 -
p.adjust:校正之后的p值 -
qvalue:q值 -
geneID:样本基因中富集到这个KEGG通路上的具体基因EntrezID,注意不是基因的symbol/Ensembl -
count:样本基因中富集到这个KEGG通路上的基因的数目,即GeneRatio的分子
2种可视化图:
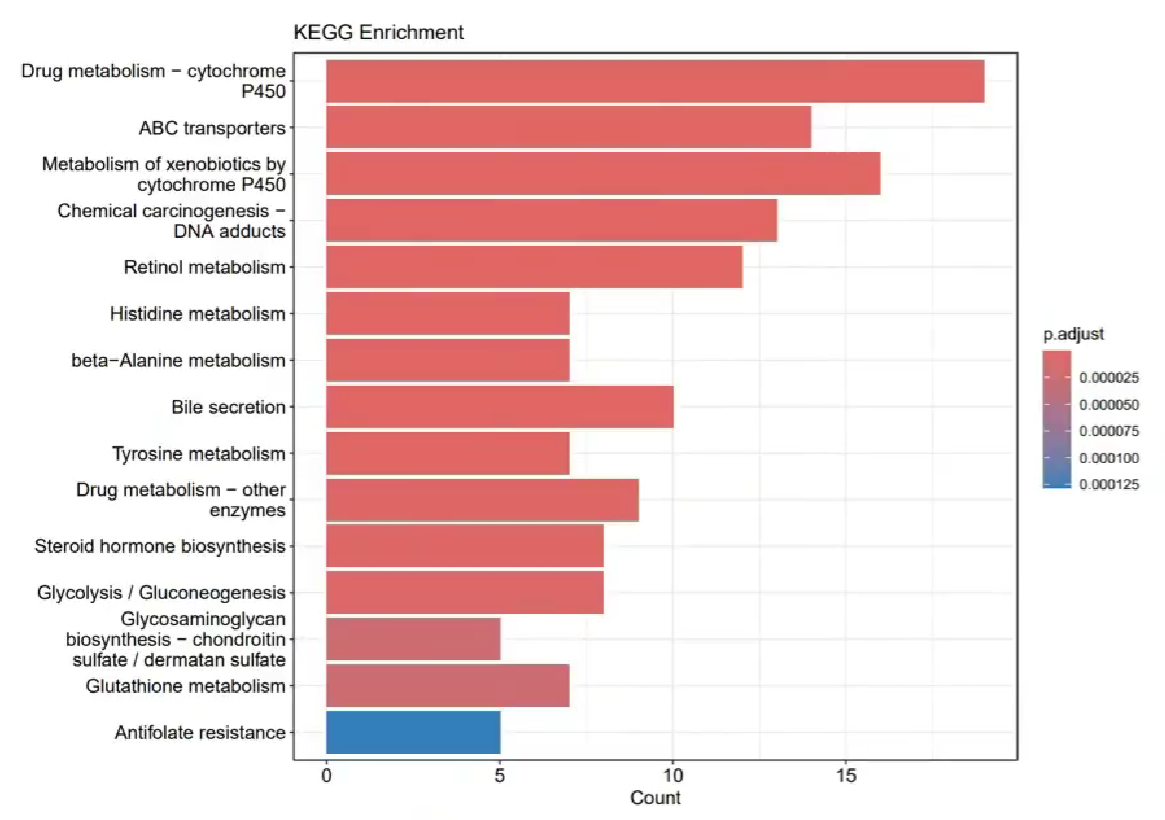
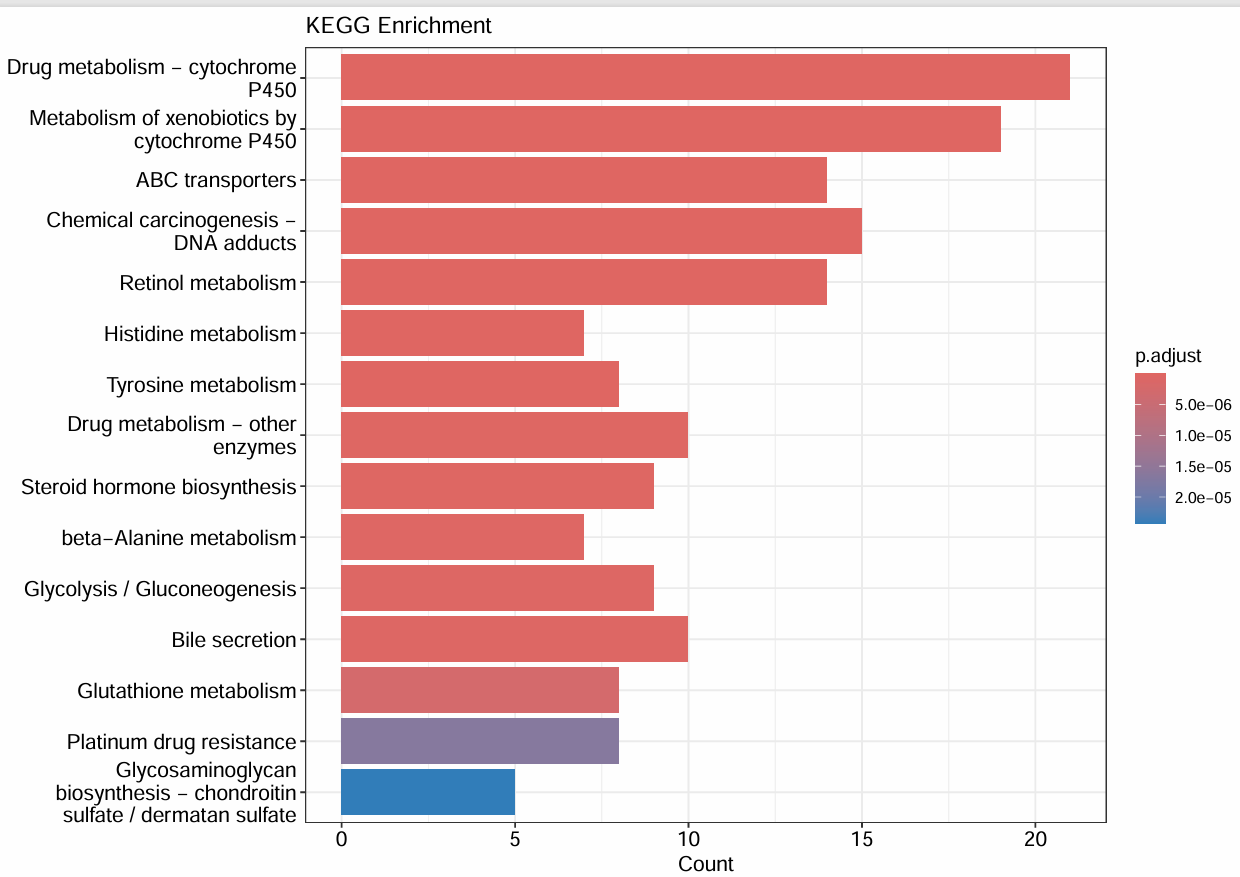
纵轴是KEGG通路的名称,横轴是富集到某个通路的基因数量,柱子的颜色是p值,越红则越小
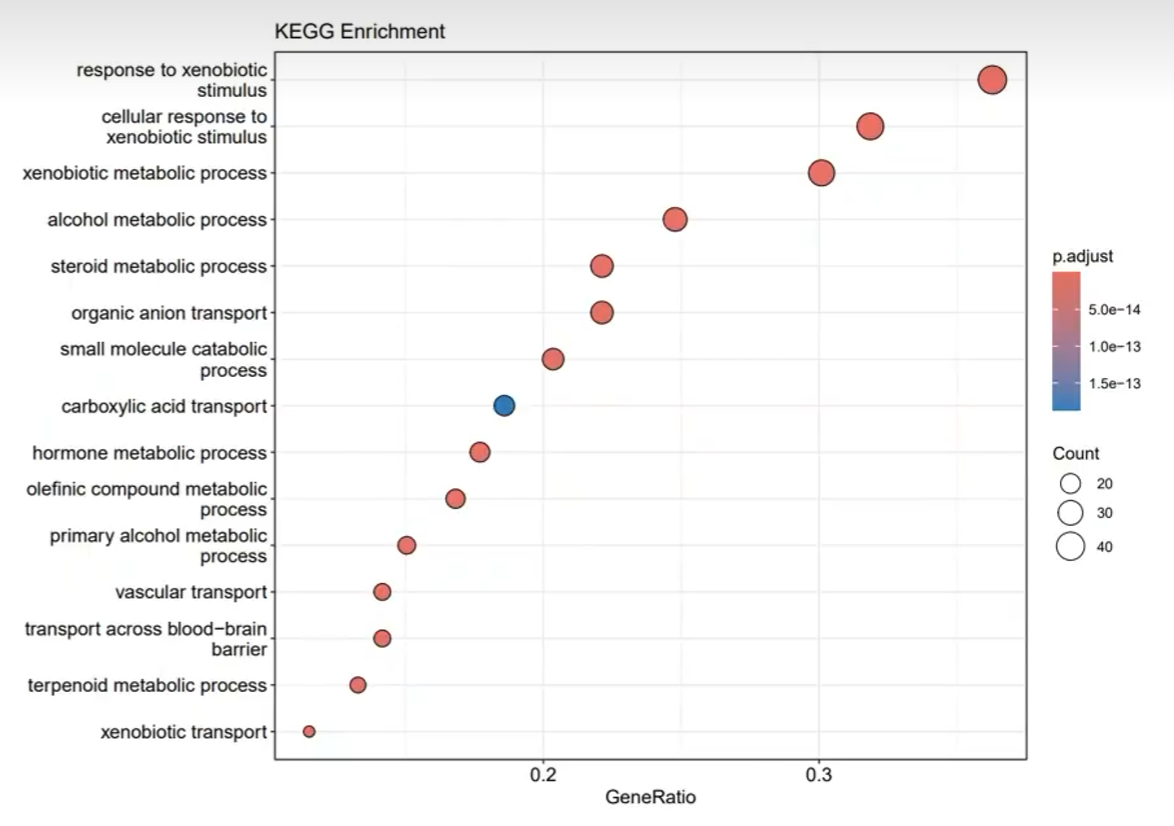
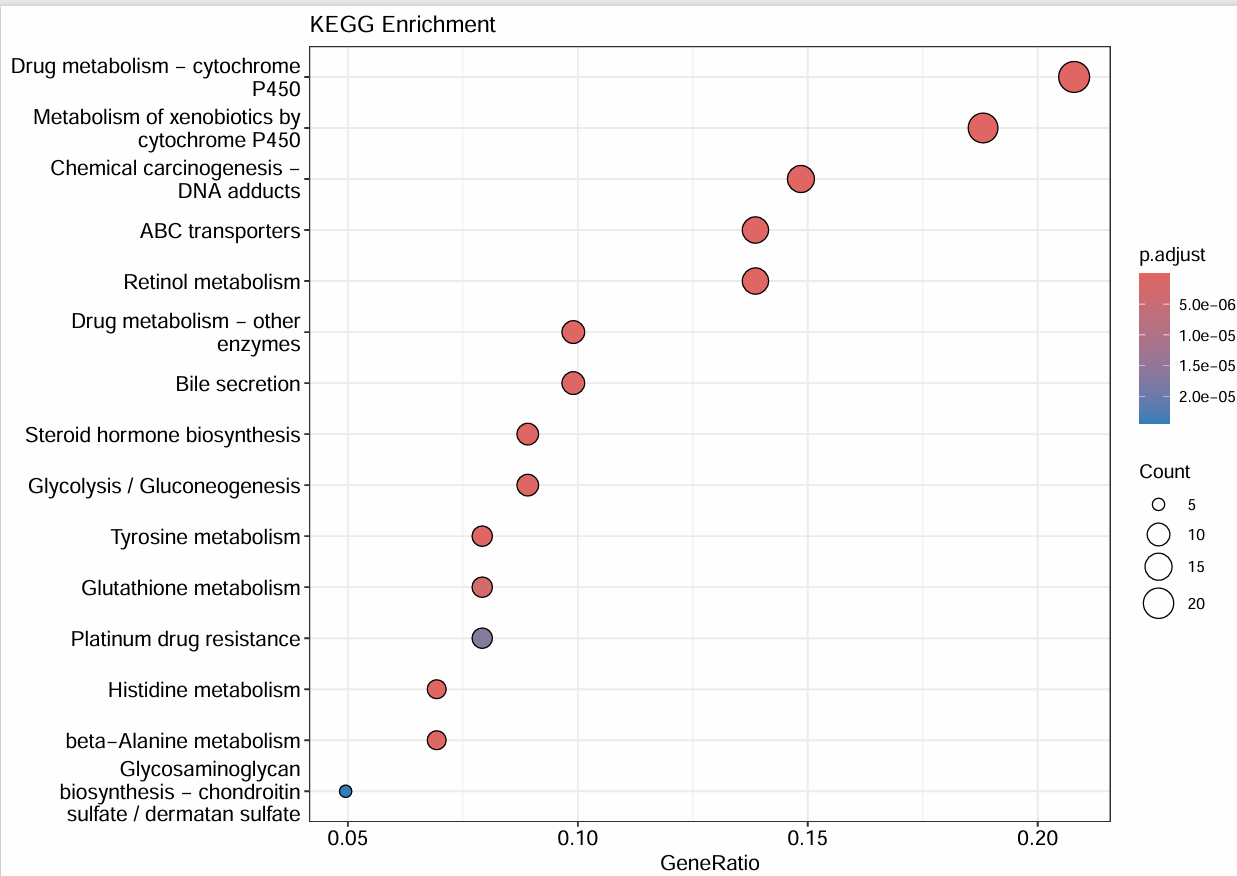
纵轴是KEGG通路的名称,横轴是GeneRatio,圆点的大小代表富集到某个通路的基因数量,圆点的颜色是p值,越红则越小
进行分析
还是使用clusterProfiler包和org.Hs.eg.db包
library("clusterProfiler");
library("org.Hs.eg.db");
load("data\\special_gene.rda");
前面提到,KEGG分析只能使用基因的EntrezID,而之前得到的都是基因的symbol,所以需要进行转换
entrezid <- mapIds(
x = org.Hs.eg.db, # 使用的数据库
keys = special_gene, # 输入的数据
keytype = "SYMBOL", # 输入数据的类型
column = "ENTREZID" # 输出数据的类型(要转成哪种数据)
);
view(entrezid);
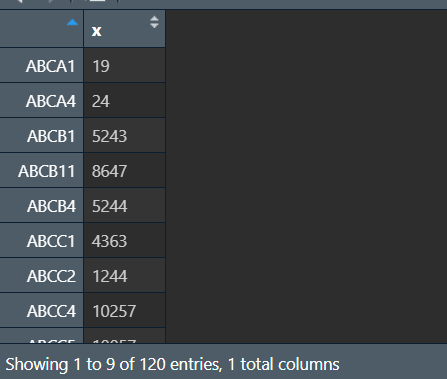
结果中x列即为基因的EntrezID
接下来使用enrichKEGG函数进行KEGG分析,注意该过程需要联网,因为是到KEGG网站上实时搜索
enrich.kegg <- enrichKEGG(
gene = entrezid, # 要进行分析的基因
organism = "hsa", # 富集物种为人类
pAdjustMethod = "BH", # 指定用FDR为p值校正方法
pvalueCutoff = 0.05, # 指定p值阈值<0.05
qvalueCutoff = 0.2 # 指定q值阈值<0.2
);
kegg_df <- as.data.frame(enrich.kegg);
view(kegg_df);
# 存储数据
write.csv(kegg_df, "data\\kegg_enrich.csv", row.names = F);
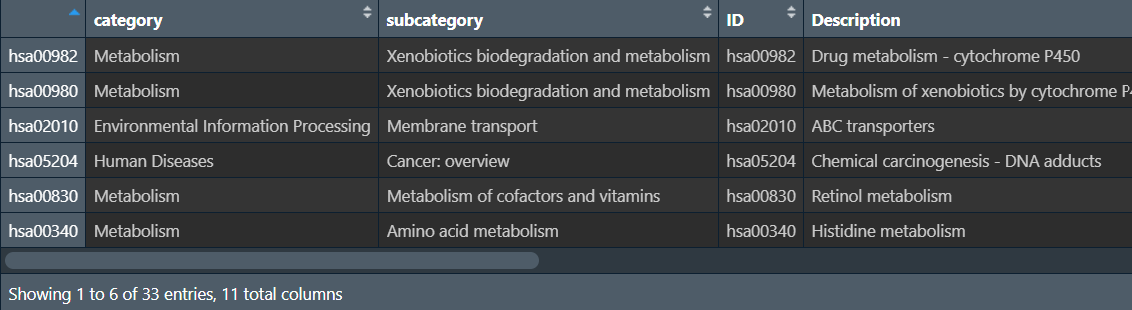
关于富集物种的设定,更多物种缩写
富集分析图
作图流程与GO富集分析类似,都是先创建(打开)一个PDF文件,绘图,关闭文件
第一个图:
pdf(file = "data\\KEGG_bar.pdf", width = 10);
barplot(enrich.kegg, # 使用enrich.kegg数据
showCategory = 15, # 显示前15条数据
title = "KEGG Enrichment" # 标题
);
dev.off();
第二个图:
pdf(file = "data\\KEGG_dot.pdf", width = 10);
dotplot(enrich.kegg, # 使用enrich.kegg数据
showCategory = 15, # 显示前15条数据
title = "KEGG Enrichment" # 标题
);
dev.off();
KEGG通路图
如果我们想单独详解某个通路,可以到KEGG官网上搜索,也可以用R来画
使用pathview包
if(!require("pathview", quietly = T))
{
library("BiocManager");
BiocManager::install("pathview");
library("pathview");
}
以KEGG结果中的hsa00982为例,注意该过程还需联网
hsa00982 <- pathview(
gene.data = special_gene, # 输入的基因
pathway.id = "hsa00982", # 通路名称
species = "hsa", # 物种
gene.idtype = "SYMBOL", # 输入基因的类型,三种都可以
kegg.native = T # 输出png图片(TRUE)或pdf文件(FALSE)
);
结果保存在r文件目录下的hsa00982.png中
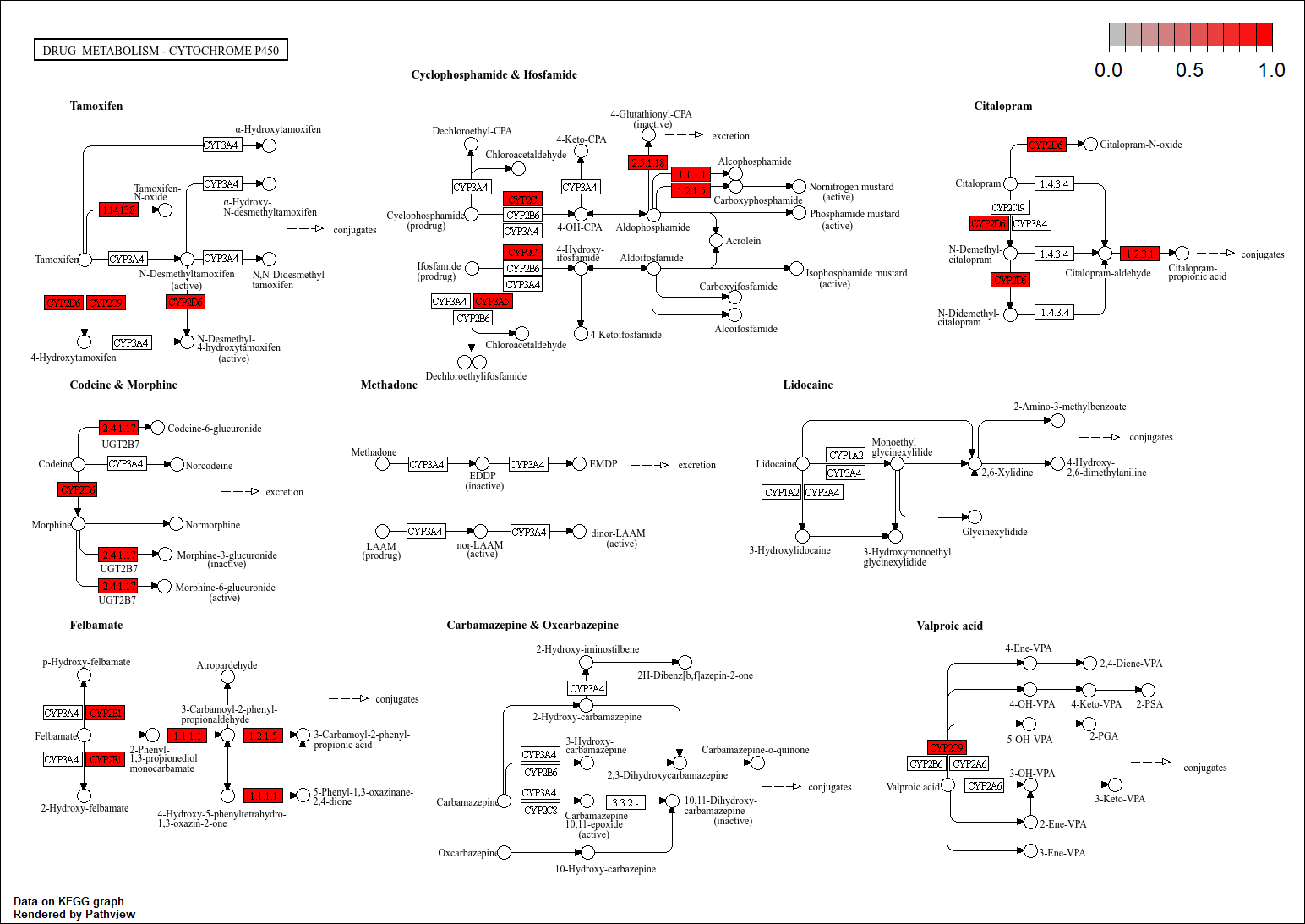
图中标红的即为special_gene中的哪些基因出现在该通路上
还可以进一步处理这个图:在我们的项目中,在这个通路上出现差异化表达的基因的表达水平是上升还是下调,可以把基因上升/下调的水平放在通路上体现出来
load("data\\resSig.rda");
deg_kegg1 <- resSig[special_gene, ]; # 要进行分析的差异化基因
deg_kegg2 <- deg_kegg1$log2FoldChange; # 差异化表达的程度
names(deg_kegg2) <- special_gene; # 上步取完后没有行名(基因名),需要重新将基因名添加为行名
view(deg_kegg2);
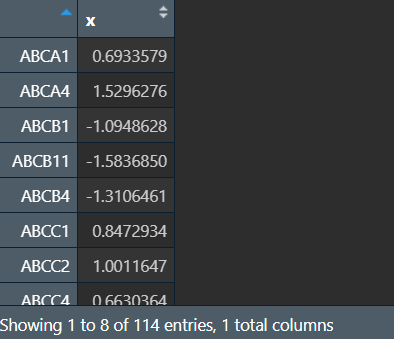
(因这部分重新更新了数据,special_gene的数量与原来不同,正常deg_kegg2的行数应与special_gene相同)
仍然用pathview函数,deg_kegg2中包含log2FoldChange和基因名两个信息,相比只有基因名的special_gene,作出的图更详细
hsa00982_new <- pathview(
gene.data = deg_kegg2, # 与上面的相同,只是使用的数据不同
pathway.id = "hsa00982",
species = "hsa",
gene.idtype = "SYMBOL",
kegg.native = T,
limit = list(gene = max(abs(deg_kegg2)), cpd = 1) # 图例
);
结果保存在r文件目录下的hsa00982.pathview.png中
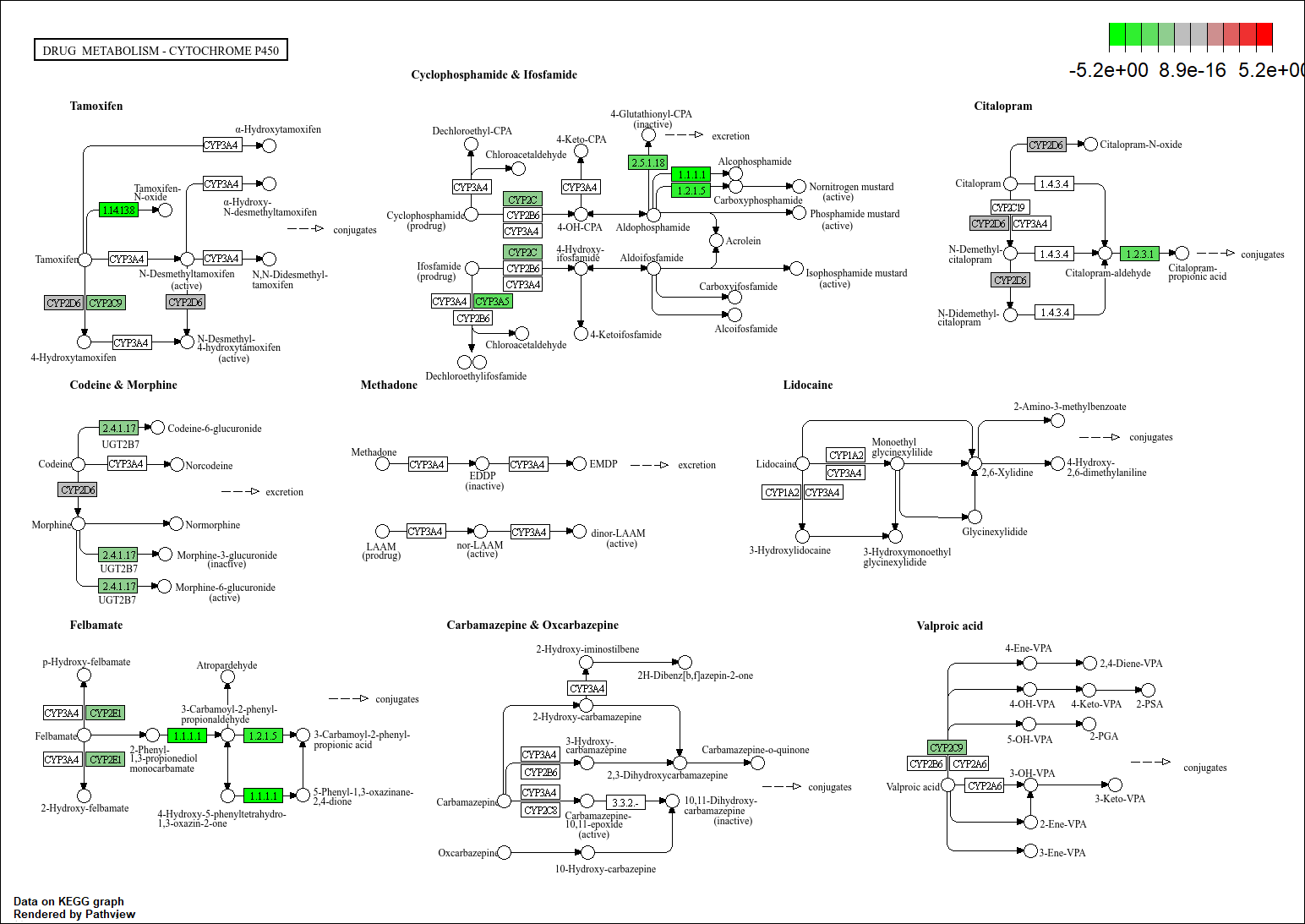
在图中,绿色代表表达下降,红色代表上升,颜色越深程度越高
代码汇总
rm(list=ls());
library("clusterProfiler");
library("org.Hs.eg.db");
load("data\\special_gene.rda");
entrezid <- mapIds(
x = org.Hs.eg.db,
keys = special_gene,
keytype = "SYMBOL",
column = "ENTREZID"
);
enrich.kegg <- enrichKEGG(
gene = entrezid,
organism = "hsa",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.2
);
kegg_df <- as.data.frame(enrich.kegg);
# 存储数据
write.csv(kegg_df, "data\\kegg_enrich.csv", row.names = F);
# 第一个KEGG富集图
pdf(file = "data\\KEGG_bar.pdf", width = 10);
barplot(enrich.kegg, showCategory = 15, title = "KEGG Enrichment");
dev.off();
# 第二个KEGG富集图
pdf(file = "data\\KEGG_dot.pdf", width = 10);
dotplot(enrich.kegg, showCategory = 15, title = "KEGG Enrichment");
dev.off();
# 第一个KEGG通路图
if(!require("pathview", quietly = T))
{
library("BiocManager");
BiocManager::install("pathview");
library("pathview");
}
hsa00982 <- pathview(
gene.data = special_gene,
pathway.id = "hsa00982",
species = "hsa",
gene.idtype = "SYMBOL",
kegg.native = T
);
# 第二个KEGG通路图
load("data\\resSig.rda");
deg_kegg1 <- resSig[special_gene, ];
deg_kegg2 <- deg_kegg1$log2FoldChange;
names(deg_kegg2) <- special_gene;
hsa00982_new <- pathview(
gene.data = deg_kegg2,
pathway.id = "hsa00982",
species = "hsa",
gene.idtype = "SYMBOL",
kegg.native = T,
limit = list(gene = max(abs(deg_kegg2)), cpd = 1)
);
PPI网络图
基础概念
在找到差异表达基因后,可以检索编码蛋白间可能的潜在相互作用,并构建蛋白质相互作用网络。目的是描述这些基因或蛋白之间存在怎样的相互关系(例如物理接触、靶向调节等),最终阐述生物体中有意义的分子调节网络
简单来说,就是找到各蛋白的相互关系
string官网的简单使用
得到special_gene的csv格式文件
rm(list=ls());
load("data\\special_gene.rda");
library("readr");
write_csv(as.data.frame(special_gene), "data\\special_gene.csv");
使用excel打开得到的csv文件,选中全部的差异基因(注意不复制第一行的special_gene列名),复制
进入string数据库官网,点击首页的search,在右侧栏中选择Multiple proteins,粘贴,物种Organisms选择人类Homo sapiens,点击search进行搜索
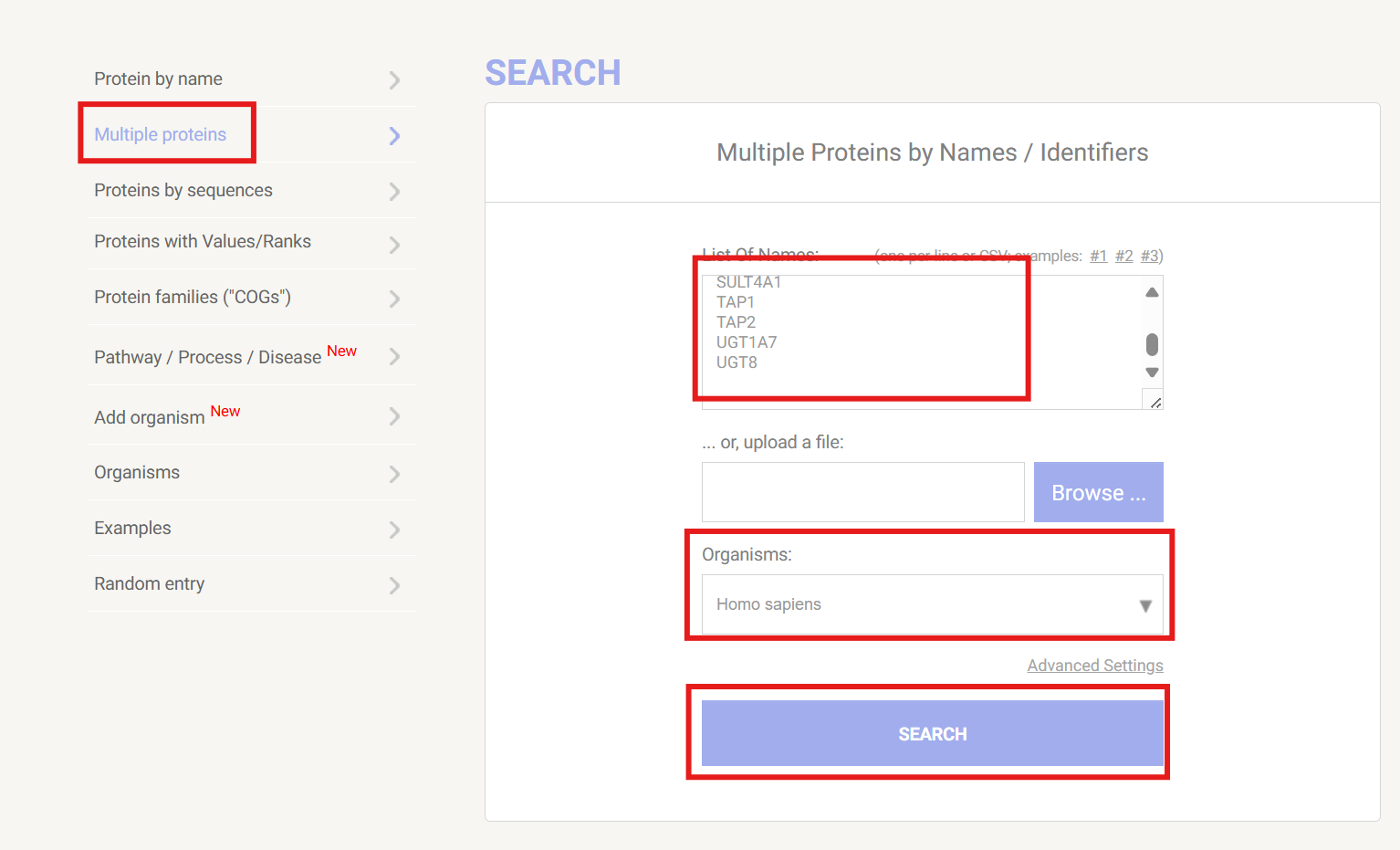
这里可以检查一下每个基因自动搜索出的结果是不是与输入的基因匹配,如果最后画出来的图与差异基因不一致,可能是这里的问题
之后点击continue
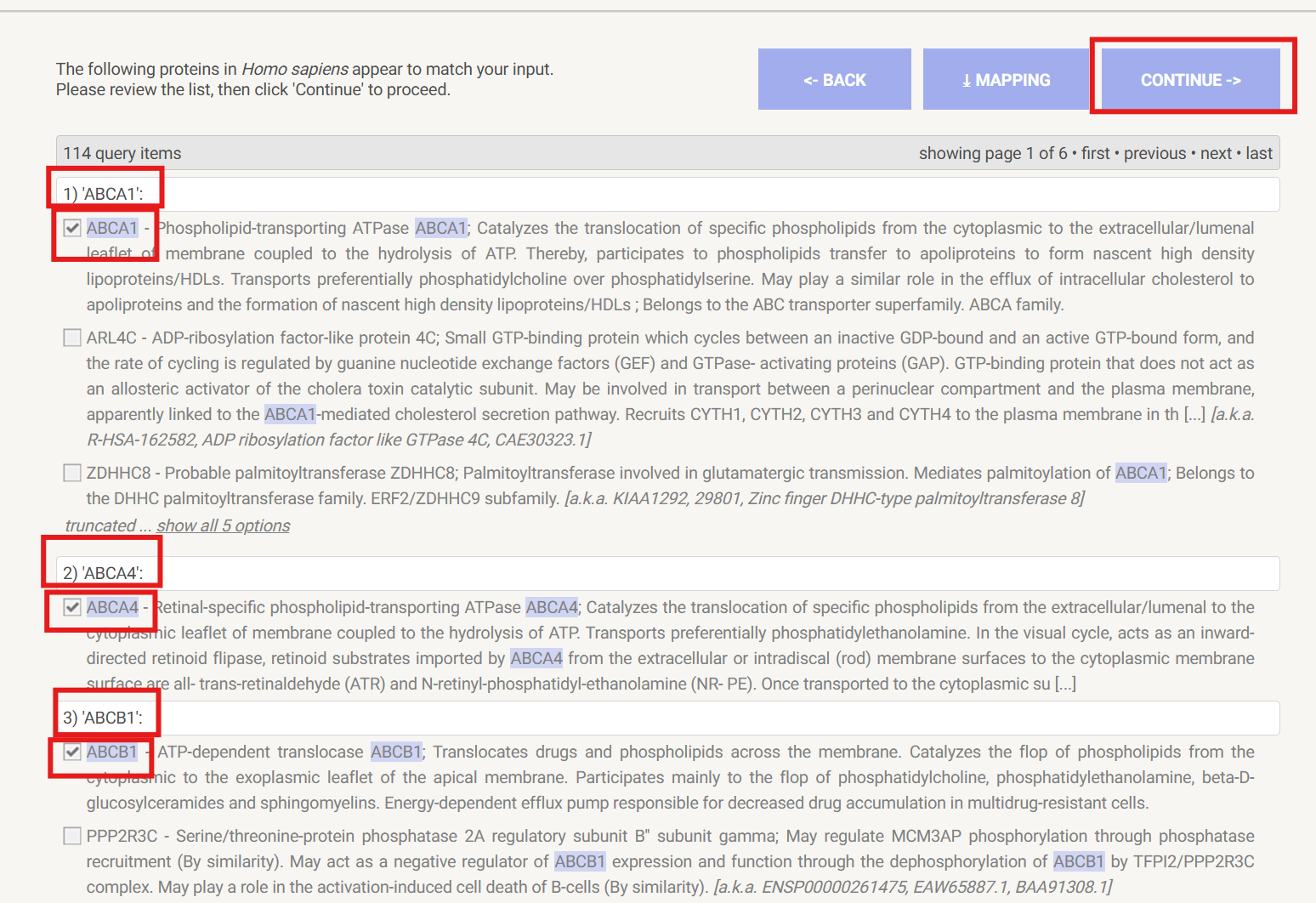
就可以画出基本的图

可以看到这里画出的图很凌乱,且有一些不紧密的相互作用可以忽略掉
如果我们觉得图中的蛋白分组太多了,可以点击Settings,调高得分置信度minimum required interaction score(这里调到了0.7);还可以在底下的network display options中点击hide disconnected nodes in the network隐藏没有连接的节点
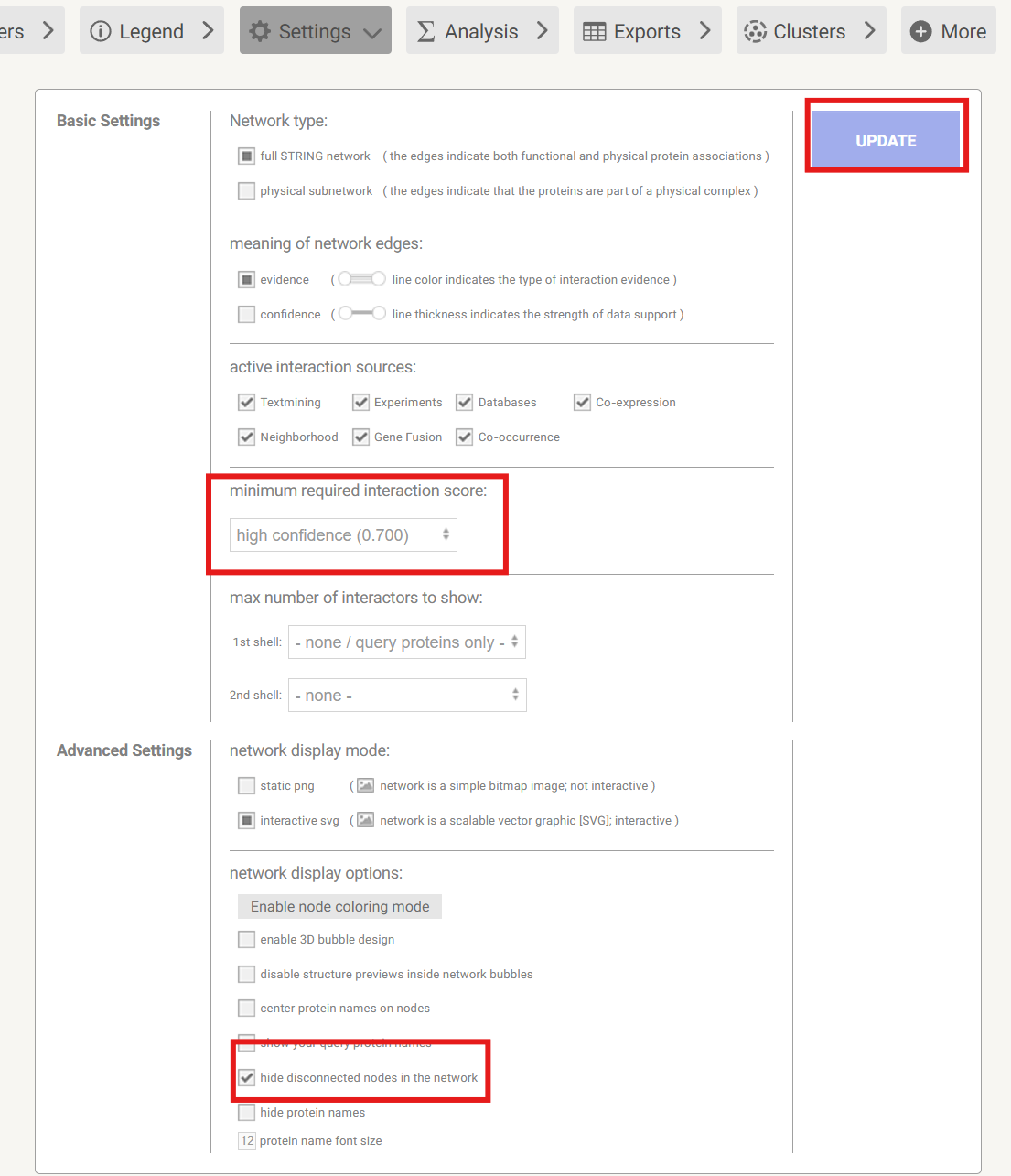
点击UPDATE更新画出的图
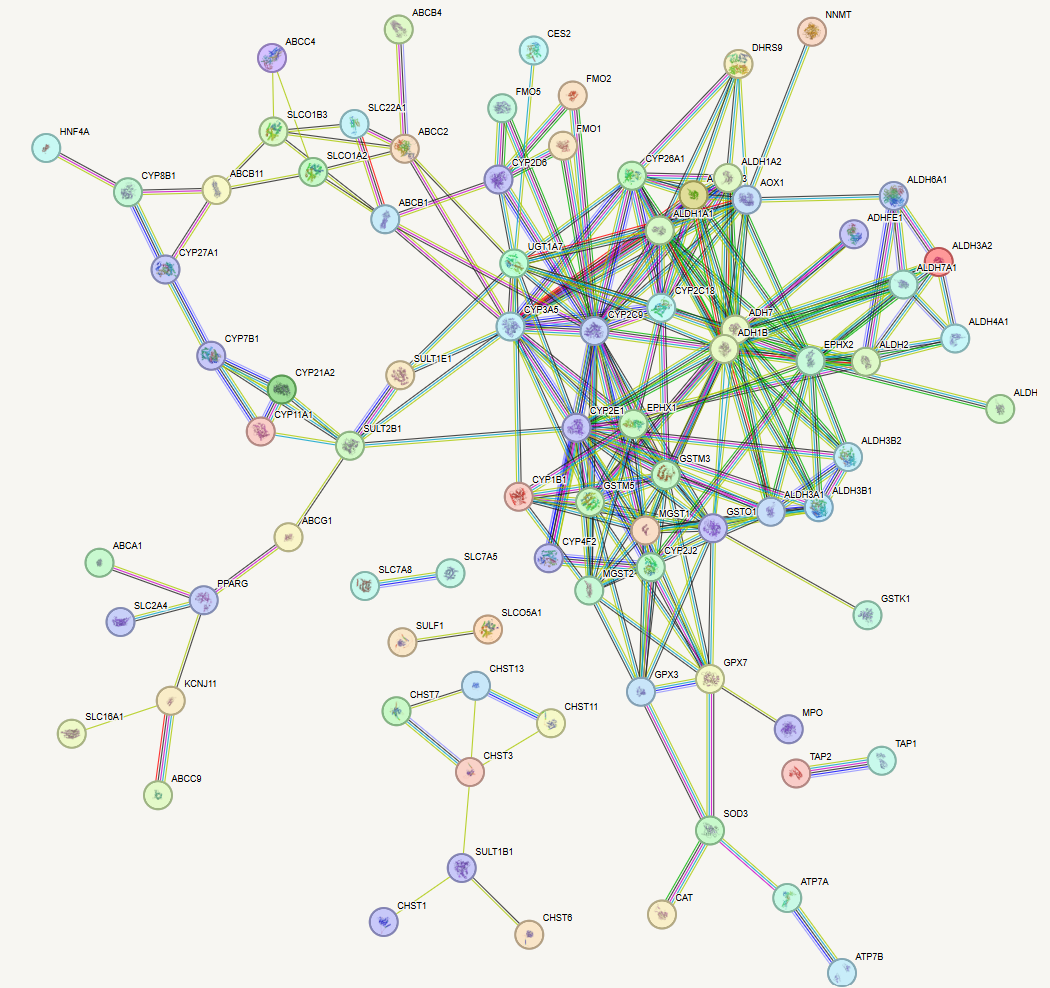
但这个图还是比较凌乱,虽然可以看到某些个分子的联系明显比其它的多,但无法直观去判断有哪些是较多的
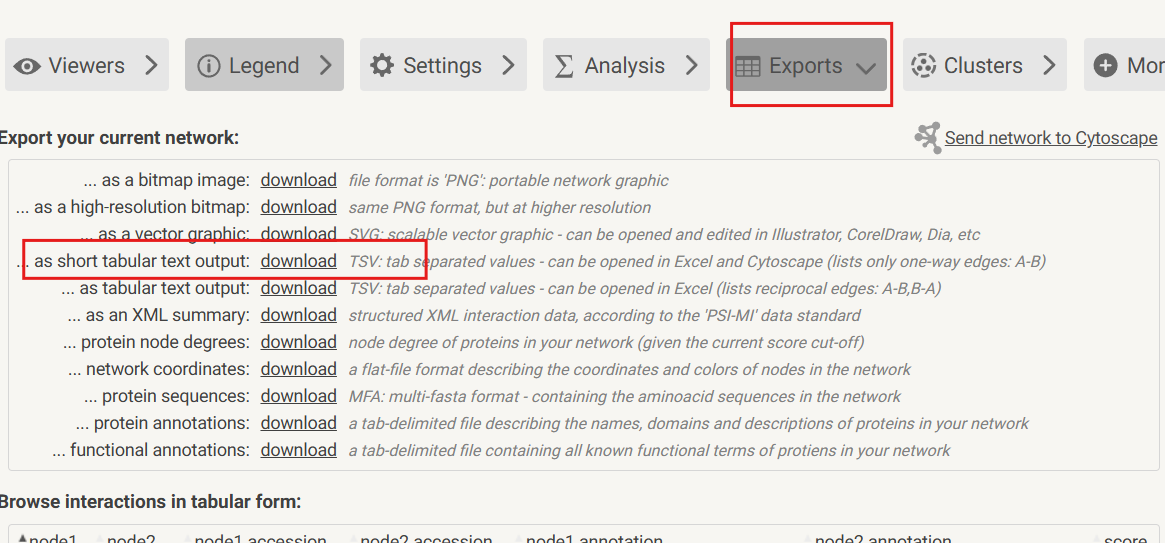
解决方法:将数据导出,去Cytoscape中分析
点击Exports,导出成TSV格式:
Cytoscape
直接在Cytoscape中打开刚才下载的文件
点击Tools->Analyze Network->OK
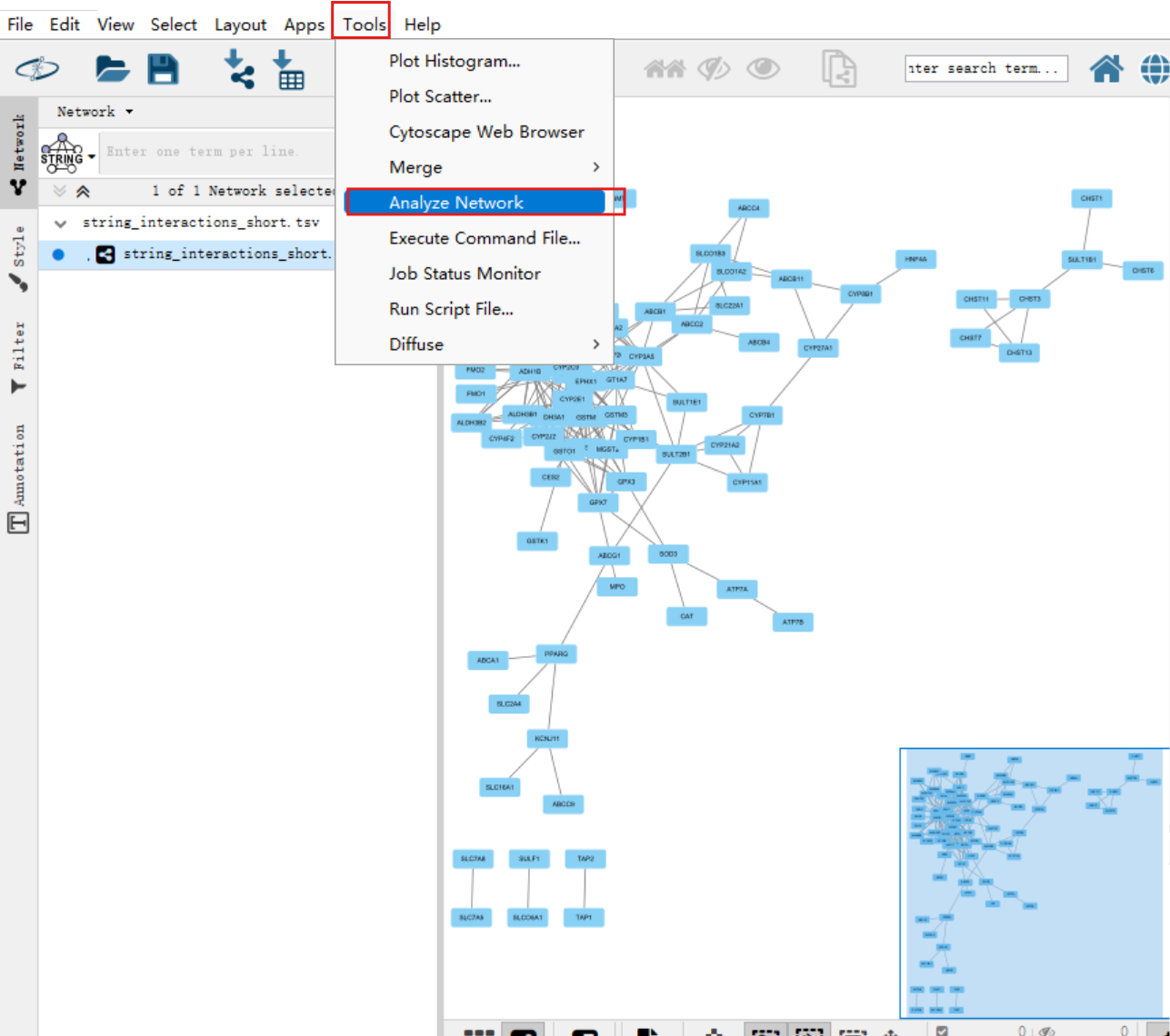
得到结果如下:
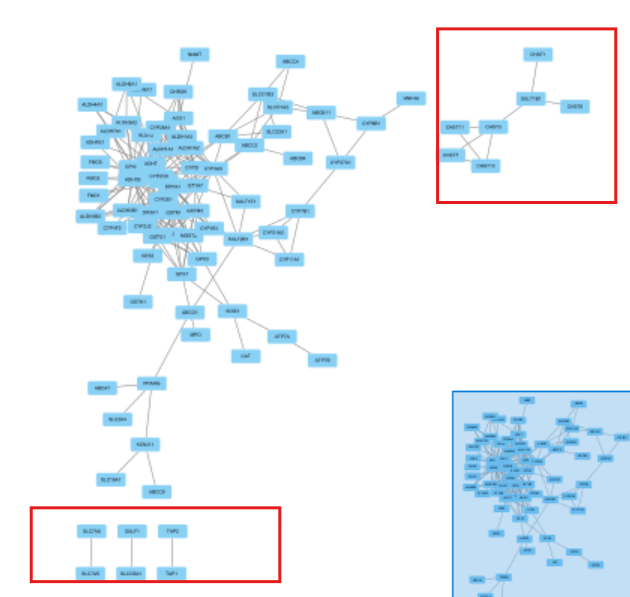
可以看到有一些基因与主干脱离,可以删去,按住CTRL,移动鼠标选中,Delete

修改节点样式:点击左侧栏中的Style,将用长方形表示的节点换成圆形的,再点击Lock node width and height,就可以将椭圆变正圆
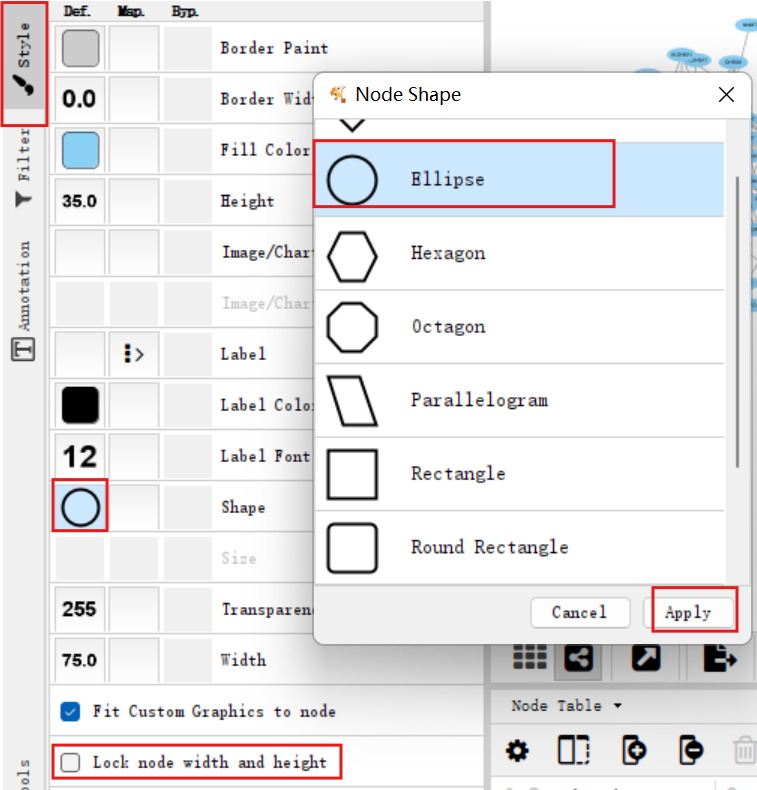
之后根据每个节点与其它节点联系数的多少给节点修改颜色,越深则越多:设置fill color的column为”degree”、mapping type为”continuous mapping”,双击current mapping选择另一种色盘
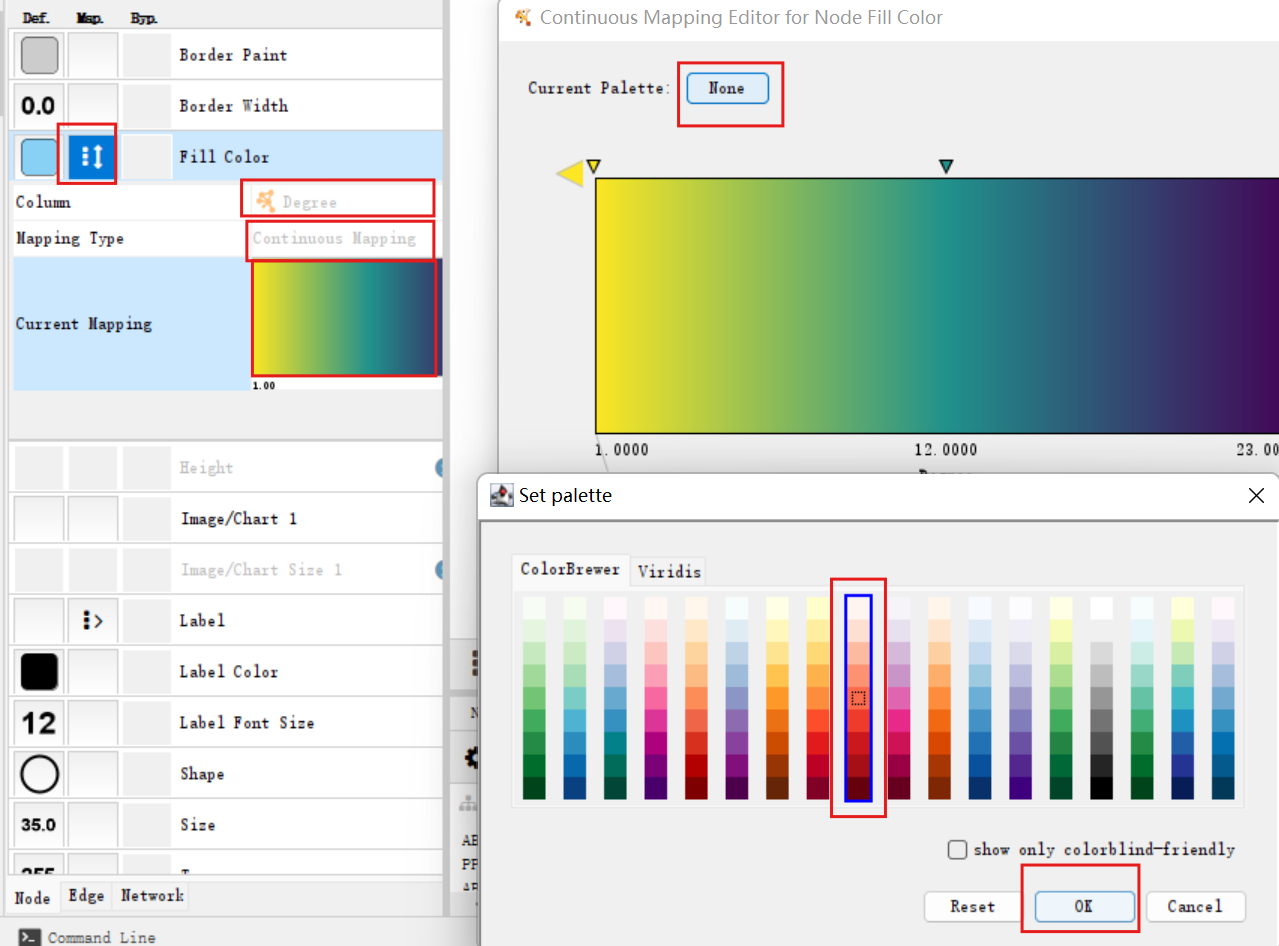
还可以把节点的大小也按联系数排列:设置size的column为”degree”、mapping type为”continuous mapping”,,双击current mapping设定最小的节点size为30,最大的设定为80
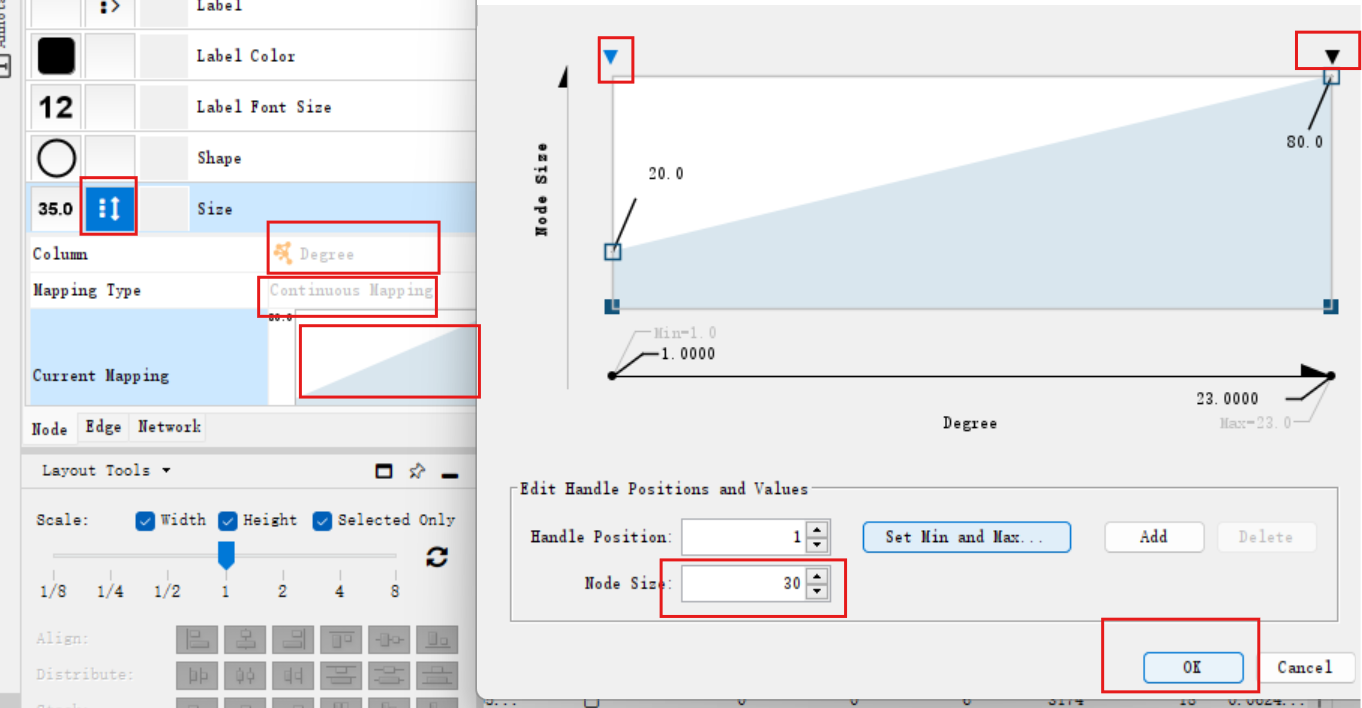
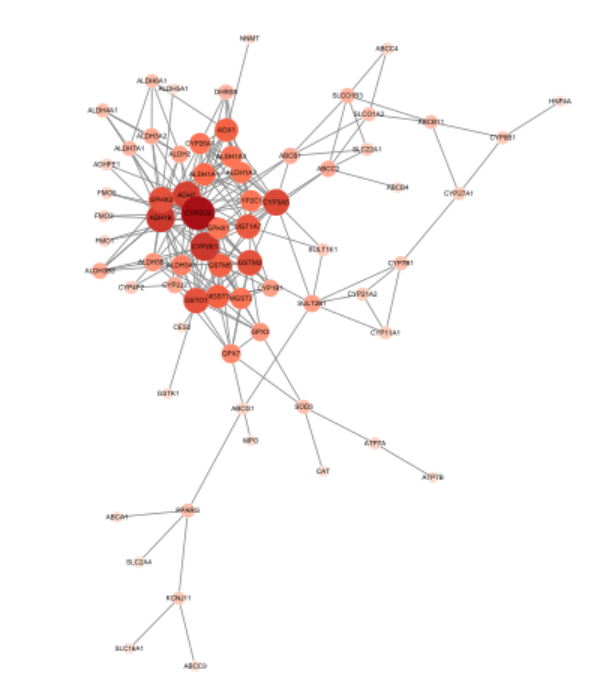
现在的图就可以很清晰的显示联系多少,但排列很杂乱
改变排列方式:将图下面的点显示方式改成Node Table,并按degree(联系数)从大到小排列,右键点击degree最大的那个节点,点击select nodes from selected rows选中它
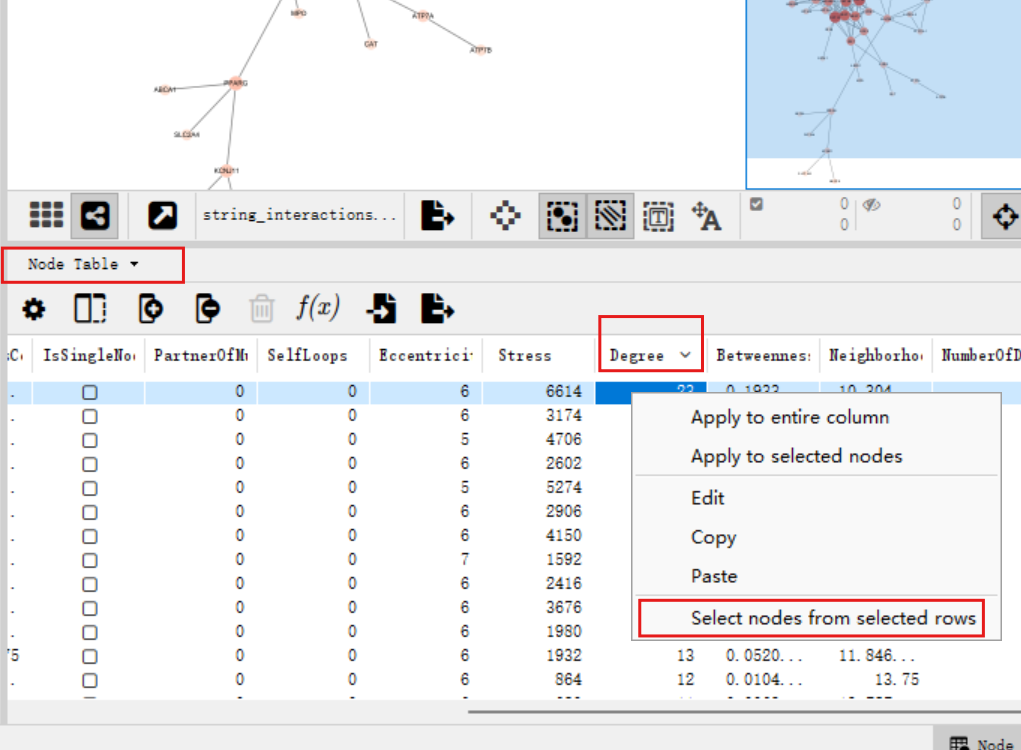
在上面的图中将它拖出来,之后单击空白处取消选中
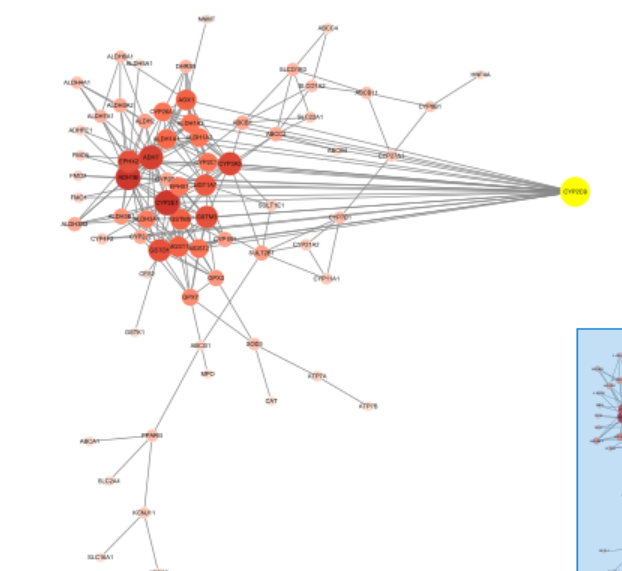
再在底下选中degree>=15的其它全部节点,右键select nodes from selected rows选中(可以单击第2行,再按住shift点击degree=15的节点,就可以选中它们之间的全部行)
点击上边栏中的layout->attribute circle layout->selected nodes only->selected
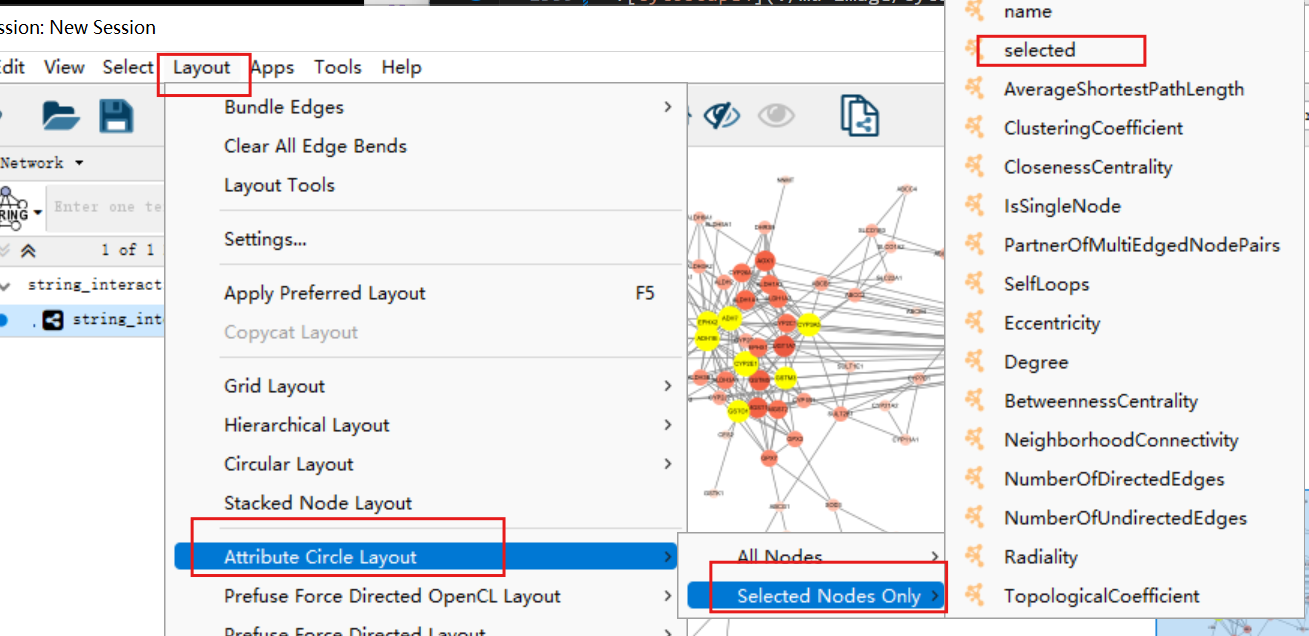
将它们拖出来,围绕着之前选出来的最高degree节点:
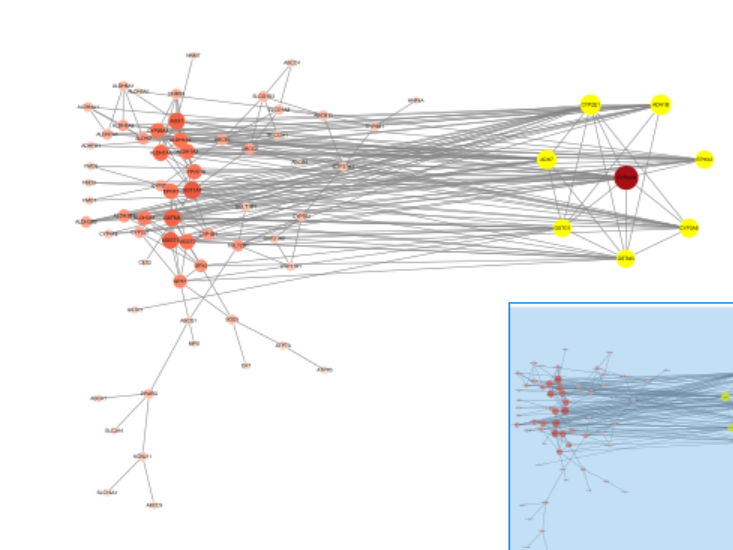
这就是第一个圆环
同样方法我们选中degree从14-6的节点为第二环:
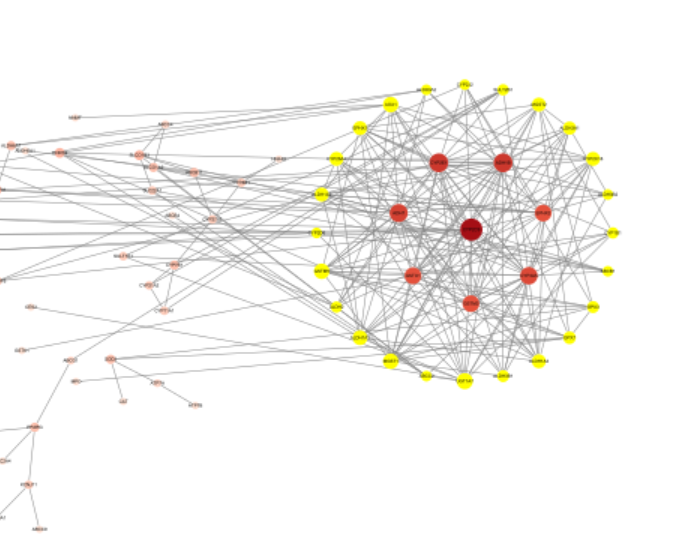
第三环是剩余的节点:
这里的圆环可能会出现排列不均的问题,如第二环与第三环过近、第三环与第四环过远
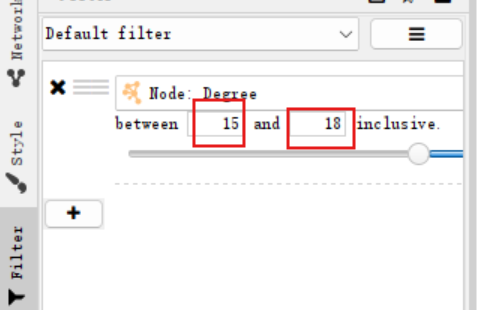
调整圆环排列:点击右侧栏中的filter,新建一个column filter
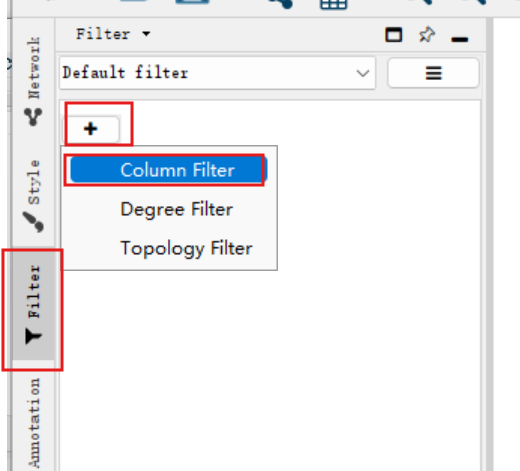
按degree进行选择:
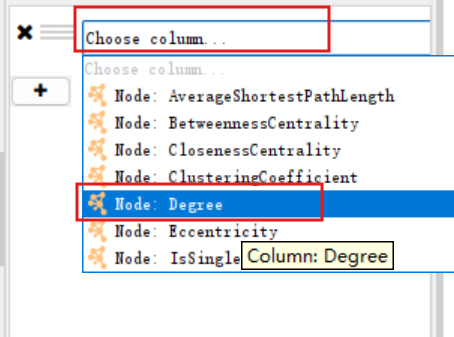
假设我们想调整第一环,即degree从18-15的节点,就输入between 15 and 18 inclusive
点击上边栏的layout->layout tools,在下面调整scale半径的滚动条至合适位置:
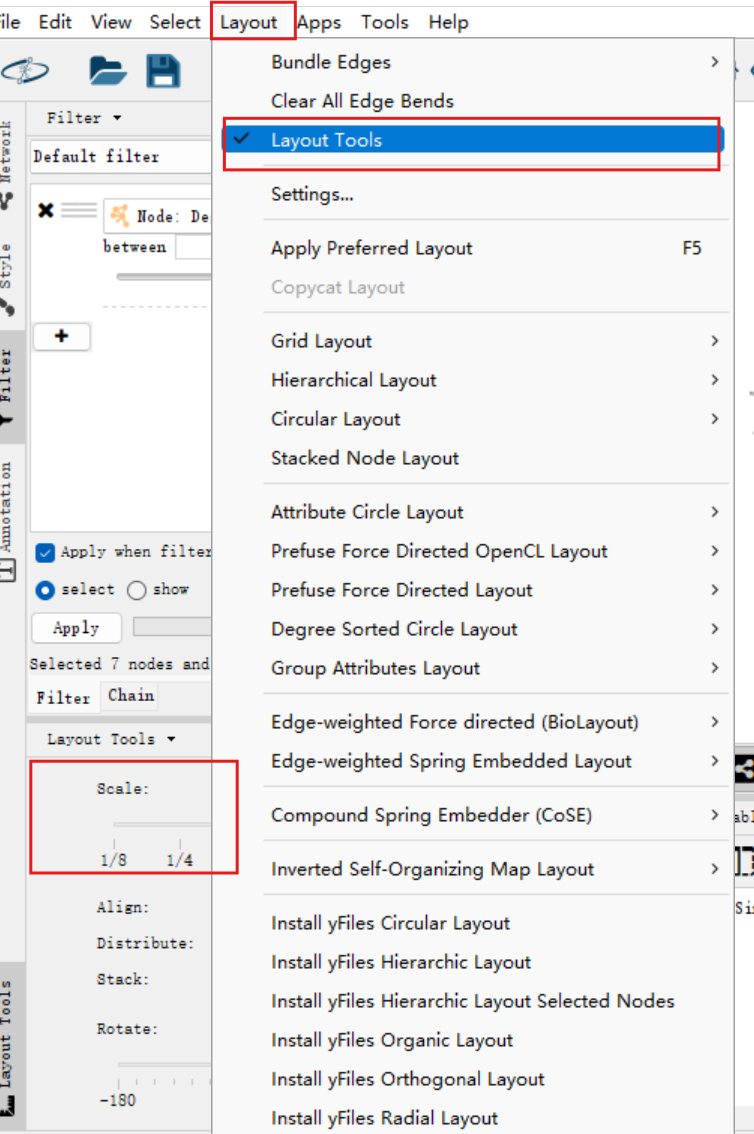
同样方式再调整其它的环
因为我这里所有环位置都还可以,就不作调整
其它的如字号(label font size)、字体颜色(label color)、节点边框和宽度(border paint/width)、透明度(transparency)都可以自己去调整
导出:点击上边栏的file->export->network to image
一般情况下都导出为PDF
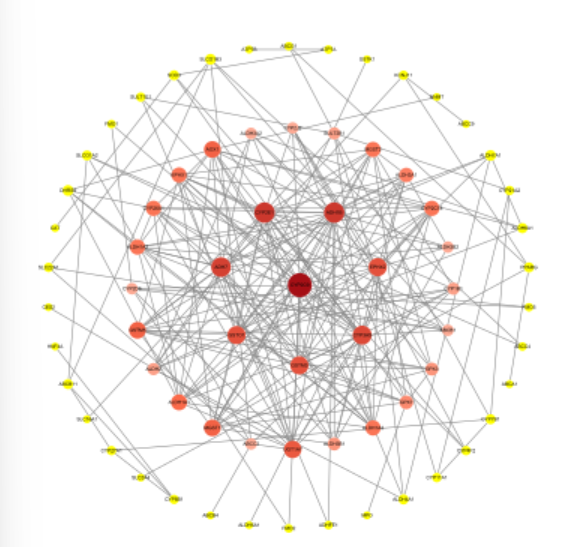
最终得到的PDF如下:

单因素Cox回归分析
概念介绍
前文单因素cox分析已经详细介绍过,这里只作简单总结
解决两个问题:哪类群体的”死亡”速度更快、什么因素影响了”死亡”速度
需要计算每个基因的风险比HR、CI值(95%置信区间对应的HR值)、P值
-
HR>1则为风险基因,高表达促死亡;HR<1则为保护基因,高表达延缓死亡
-
CI没有跨过”1”这个点,则有统计学意义
-
P<0.05,则有统计学意义
-
当结果没有统计学意义,且HR≈1时,可以认为该基因对死亡无明显影响
一种结果图:
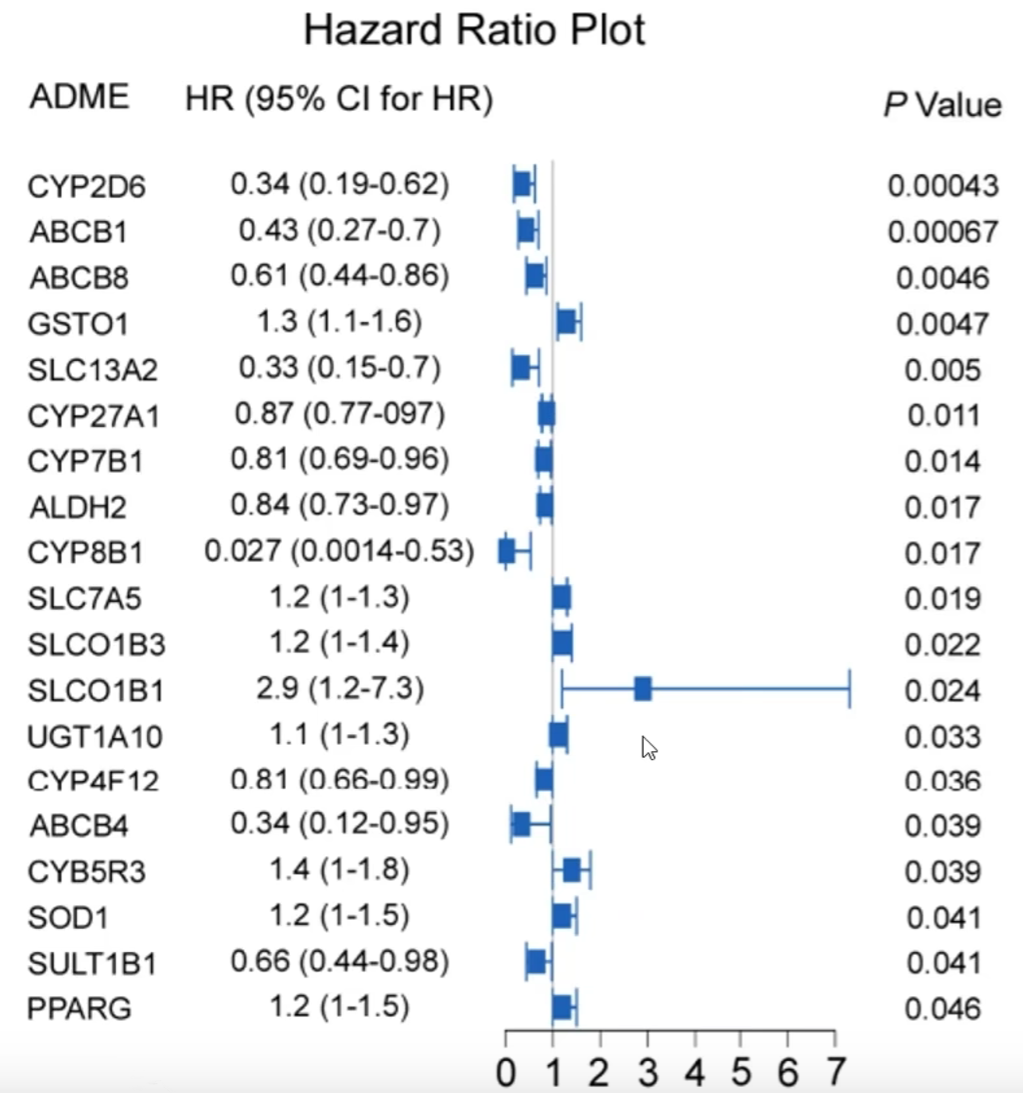
在竖线左边则HR<1,右边则>1
准备的数据:
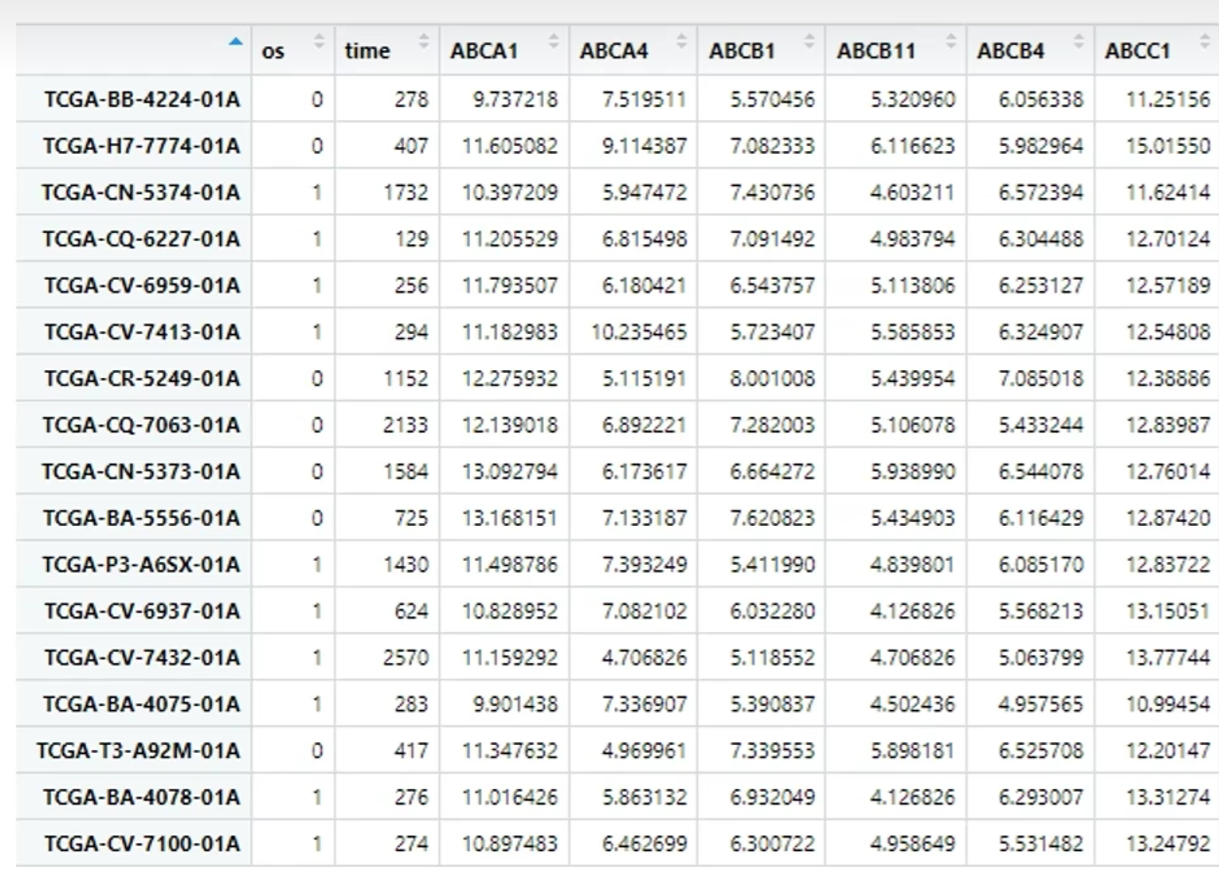
行名为样本名称,前两列分别为是否存活(0为存活,1为死亡)和存活时间(从发病到最后一次随访的时间),后面的列是基因表达量
可近似理解为:在不同样本中,各基因表达量的不同配比,在某种程度上导致了样本来源病人的不同生存时间以及结局。由此找出:具体哪些基因有利于病人的生存、哪些基因不利于病人的生存、哪些基因对病人生存没有明显影响
准备数据
载入之前得到的数据:
-
差异基因
special_gene -
差异分析结果
dds -
肿瘤组的训练组
counts_tumor_traning -
生存数据
surv_data
load("data\\special_gene.rda");
load("data\\dds.rda");
load("data\\counts_tumor_traning.rda");
load("data\\surv_data.rda");
vst标准化转换:
library("DESeq2");
rld <- vst(dds, blind=T); # 对差异分析结果进行vst标准化转换
expr_norm <- assay(rld); # 转换为矩阵形式
获取差异化表达的、肿瘤组的基因表达数据:
expr_for_cox <- expr_norm[special_gene,]; # 筛选出差异化表达基因
expr_for_cox_tumor <- expr_for_cox[, colnames(counts_tumor_traning)]; # 仅保留肿瘤组
expr_for_cox_tumor_t <- t(expr_for_cox_tumor); # 转置,便于后续数据合并
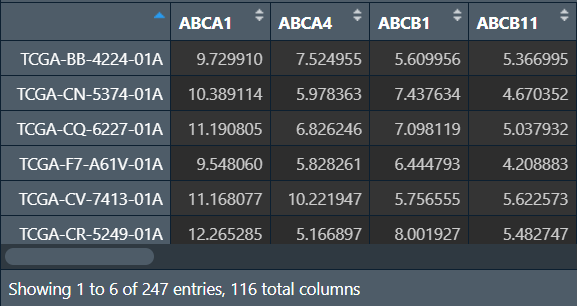
这样样本名就是行名,基因名就是列名
获取肿瘤组样本的生存数据:
surv_for_cox <- surv_data[colnames(counts_tumor_traning), ]; # 肿瘤组生存数据
surv_for_cox <- surv_for_cox[, -2]; # 仅保留OS、OS.TIME两列
colnames(surv_for_cox) <- c("os", "time"); # 改列名
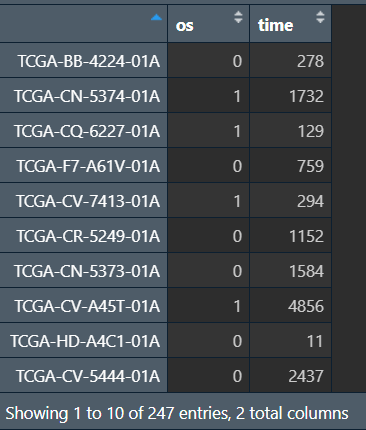
最后只需将两组数据列合并即可:
dat_cox <- cbind(surv_for_cox, expr_for_cox_tumor_t); # 合并数据
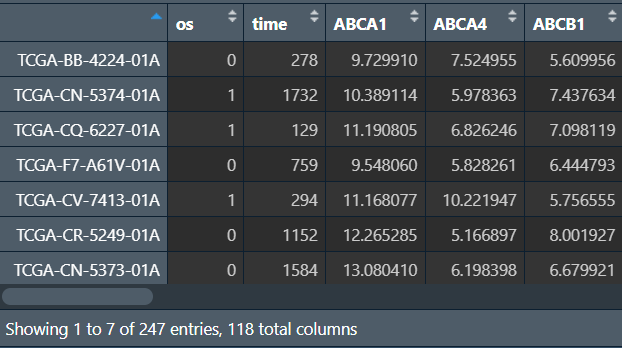
共118列,为surv_for_cox的2列(os和time列)+expr_for_cox_tumor_t的116列(116个差异基因)
这样就得到了与前面相同的数据
进行分析
cox分析函数:
Cox_uni <- function(dat, gene_list){ # dat即为上面得到的数据,gene_list是特异表达基因
library("survival");
gene_list <- gsub(gene_list, pattern = '-', replacement = '_'); # 调整gene_list格式
uni_cox <- function(single_gene){ # 真正对每个基因进行cox分析的函数
formula <- as.formula(paste0('Surv(time, os)~', single_gene)); # 公式
cox <- coxph(formula, data = dat) # cox分析函数
sum_cox <- summary(cox); # 对分析结果进行总结
if(sum_cox$coefficients[, 5] < 0.05){ # 如果p值小于0.05
report <- data.frame( # 对sum_cox再进行精简总结,使结果更直观
'Genes' = single_gene, # 基因名
'HR' = sum_cox$conf.int[, "exp(coef)"], # 风险比
'CI95' = paste0(round(sum_cox$conf.int[, 3:4], 2), collapse = '-'), # CI(分别取了结果中的最大最小值)
'pvalue' = sum_cox$coefficients[, 5] # p值
);
return(report); # 返回这个基因的cox分析结果,是一个只有1行的df
}
}
a <- lapply(gene_list, uni_cox);
# 对gene_list的每个元素(基因)进行cox分析,结果追加保存到a中
# a中的每个元素都是一个df且有相同的列名('Genes'、'HR'、'CI95'、'pvalue')
return(do.call(rbind, a));
# 对a中的每个元素(df)执行rbind行合并,生成所有基因的总cox分析结果
}
调用函数:
cox_uni_res <- Cox_uni(
dat = dat_cox,
gene_list = special_gene
);
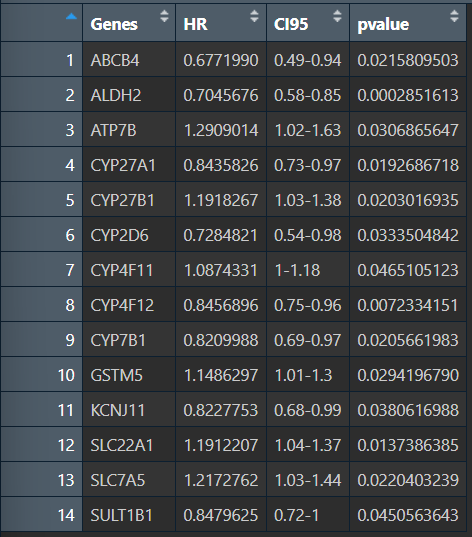
可以看到每个基因的HR值、CI区间和p值
代码汇总
为下节课准备:
-
差异化表达的、肿瘤组的基因表达数据
expr_for_cox_tumor -
用于cox分析的数据
dat_cox -
分析结果
cox_uni_res
rm(list=ls());
load("data\\special_gene.rda");
load("data\\dds.rda");
load("data\\counts_tumor_traning.rda");
load("data\\surv_data.rda");
library("DESeq2");
rld <- vst(dds, blind=T);
expr_norm <- assay(rld);
expr_for_cox <- expr_norm[special_gene,];
expr_for_cox_tumor <- expr_for_cox[, colnames(counts_tumor_traning)];
expr_for_cox_tumor_t <- t(expr_for_cox_tumor);
surv_for_cox <- surv_data[colnames(counts_tumor_traning), ];
surv_for_cox <- surv_for_cox[, -2];
colnames(surv_for_cox) <- c("os", "time");
dat_cox <- cbind(surv_for_cox, expr_for_cox_tumor_t);
Cox_uni <- function(dat, gene_list){
library("survival");
gene_list <- gsub(gene_list, pattern = '-', replacement = '_');
uni_cox <- function(single_gene){
formula <- as.formula(paste0('Surv(time, os)~', single_gene));
cox <- coxph(formula, data = dat);
sum_cox <- summary(cox);
if(sum_cox$coefficients[, 5] < 0.05){
report <- data.frame(
'Genes' = single_gene,
'HR' = sum_cox$conf.int[, "exp(coef)"],
'CI95' = paste0(round(sum_cox$conf.int[, 3:4], 2), collapse = '-'),
'pvalue' = sum_cox$coefficients[, 5]
);
return(report);
}
}
a <- lapply(gene_list, uni_cox);
return(do.call(rbind, a));
}
cox_uni_res <- Cox_uni(dat = dat_cox, gene_list = special_gene);
# 存储数据
save(expr_for_cox_tumor, file = "data\\expr_for_cox_tumor.rda");
save(dat_cox, file = "data\\dat_cox.rda");
save(cox_uni_res, file = "data\\cox_uni_res.rda");
LASSO回归
概念介绍
前文LASSO回归已经详细介绍过,这里只作简单总结
通过简化模型,丢弃一些对模型结果影响不大(可被统计学接受)的基因,挑选并保留对结果影响最大的某几个基因,作为最终形成模型的基因,此时简化后的模型与原来的模型准确度无明显差别
而LASSO回归就是挑选对结果影响最大基因的方法
准备数据
在前面的单因素Cox回归分析中,我们已经得到了对模型结果(”死亡”速度)有较大影响的几个基因,我们现在要基于这些基因进行LASSO回归
载入之前得到的数据:
-
差异化表达的、肿瘤组的基因表达数据
expr_for_cox_tumor -
用于cox分析的数据
dat_cox -
分析结果
cox_uni_res
load("data\\expr_for_cox_tumor.rda");
load("data\\dat_cox.rda");
load("data\\cox_uni_res.rda");
获取训练–肿瘤组中,cox分析得到基因的表达数据dat_cox_sur:
dat_cox_sur <- expr_for_cox_tumor[cox_uni_res$Genes, ];
# expr_for_cox_tumor是训练--肿瘤组中所有特异表达基因的表达数据
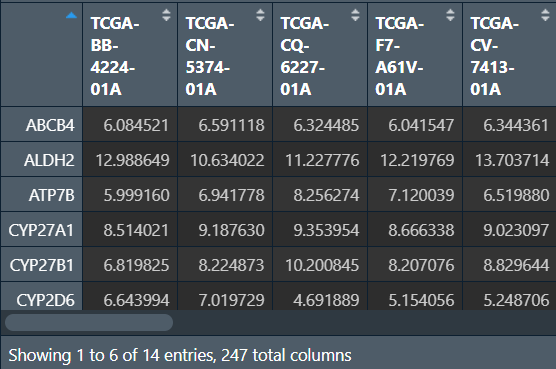
获取训练–肿瘤组中,cox分析得到基因的表达数据和生存数据dat_cox_2:
library("tidyverse");
dat_cox_2 <- dat_cox %>%
# dat_cox是训练--肿瘤组中所有特异表达基因的表达和生存数据
dplyr::select('os', 'time', cox_uni_res$Genes); # 选出os time以及cox分析基因的列
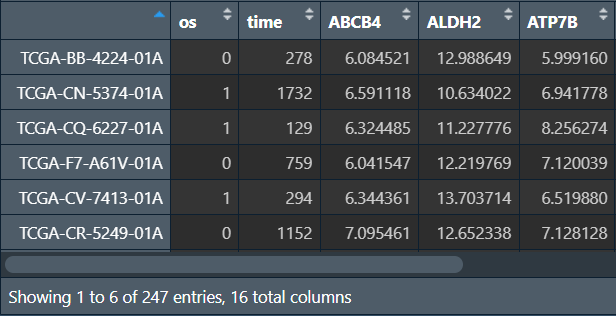
进行分析
需要glmnet包:
if(!require("glmnet", quietly = T))
{
install.packages("glmnet");
library("glmnet");
}
交叉验证曲线:
cv_fit <- cv.glmnet(
x = t(dat_cox_sur),
y = dat_cox_2$os
);
plot(cv_fit);
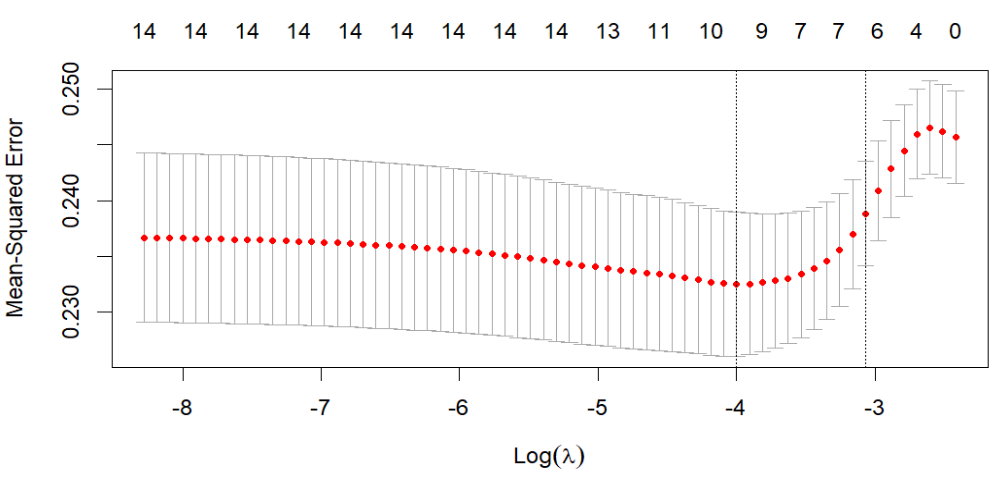
回归系数路径图:
fit <- glmnet(
x = t(dat_cox_sur),
y = dat_cox_2$os
);
plot(fit, xvar = 'lambda');
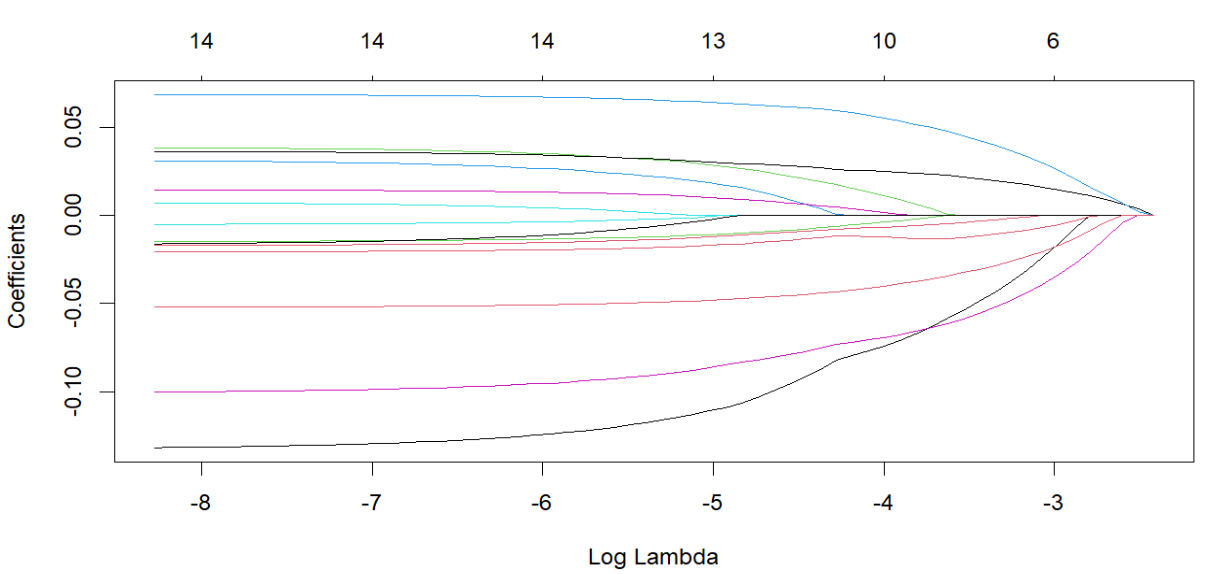
获取LASSO回归筛选出的基因:
这里我们选择偏差最小时的log λ值λ min作为结果,此时模型的拟合效果最好(当然也可以是λ1-se)
coefficient <- coef(cv_fit, s = 'lambda.min'); # 当选择λ min时的变量系数,也就是β0~βn
active.index <- which(as.numeric(coefficient) != 0); # 找到哪些变量系数不为0
active.coefficient <- as.numeric(coefficient)[active.index]; # 不为0的变量系数值
sig_gene_mult_cox <- rownames(coefficient)[active.index]; # 变量系数不为0的基因名
sig_gene_mult_cox <- sig_gene_mult_cox[-1]; # 删掉第一个值(截距β0),剩下的就是基因名了
lasso_res <- tibble( # 将基因名和其系数值整合
gene = sig_gene_mult_cox,
βn = active.coefficient[-1] # 删去β0的值
);
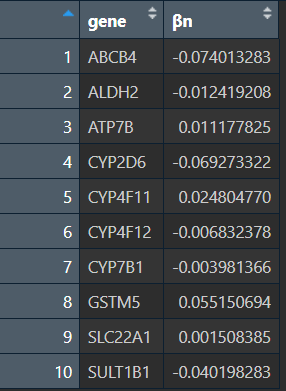
可以看到βn值小于0的基因都是延缓死亡的(HR<1),大于0的促死亡(HR>1)
代码汇总
rm(list=ls());
load("data\\expr_for_cox_tumor.rda");
load("data\\dat_cox.rda");
load("data\\cox_uni_res.rda");
dat_cox_sur <- expr_for_cox_tumor[cox_uni_res$Genes, ];
library("tidyverse");
dat_cox_2 <- dat_cox %>% dplyr::select('os', 'time', cox_uni_res$Genes);
if(!require("glmnet", quietly = T))
{
install.packages("glmnet");
library("glmnet");
}
# 交叉验证曲线
cv_fit <- cv.glmnet(x = t(dat_cox_sur), y = dat_cox_2$os);
pdf(file = "data\\cv_fit.pdf", width = 10);
plot(cv_fit);
dev.off();
# 回归系数路径图
fit <- glmnet(x = t(dat_cox_sur), y = dat_cox_2$os);
pdf(file = "data\\fit.pdf", width = 10);
plot(fit, xvar = 'lambda');
dev.off();
# lasso回归
coefficient <- coef(cv_fit, s = 'lambda.min');
active.index <- which(as.numeric(coefficient) != 0);
active.coefficient <- as.numeric(coefficient)[active.index];
sig_gene_mult_cox <- rownames(coefficient)[active.index];
sig_gene_mult_cox <- sig_gene_mult_cox[-1];
lasso_res <- tibble(gene = sig_gene_mult_cox, βn = active.coefficient[-1]);
简单总结
在完成数据预处理后,我们做了差异表达分析,得到了1万多个差异化表达基因,与ADME基因取交集后,得到100多个差异化表达的ADME基因
之后我们对它们做了GO富集分析、KEGG富集分析并画了PPI网络图
为找到对”死亡”速度有影响的基因,我们对这100多个基因进行单因素Cox回归分析,得到10多个有临床意义的差异化表达ADME基因
最后,我们为了简化模型,又对这10多个基因进行LASSO回归分析,在不大幅度影响模型准确度的基础上,再次减少了基因数量
后续无论是做预测模型、还是实验验证,都可以着重针对这些基因去做