b站生信课程02-2
r-bioinfolessonb站生信课程TCGA及GEO数据挖掘入门必看P8-P17笔记——差异表达分析、cox与lasso回归、富集分析、KM生存分析
写在前面:本篇教程来自b站课程TCGA及GEO数据挖掘入门必看 P8-P17 相关资料下载
差异表达分析(TCGA)
一般情况下,如果使用的表达矩阵是TPM值,就用wilcoxon;如果是count值,就用DESeq2等方法
TPM值
使用包:limma、pheatmap、ggVolcano
if(!require("pheatmap", quietly = T))
{
install.packages("pheatmap");
library("pheatmap");
}
if(!require("ggVolcano", quietly = T))
{
if(!require("devtools", quietly = T))
{
install.packages("devtools");
library("devtools");
}
devtools::install_github("BioSenior/ggVolcano");
library("ggVolcano");
}
library("tidyverse");
读取文件并转化为matrix,方法同前
data <- read.table(
"save_data\\TCGA_LUSC_TPM.txt",
header = T,
sep = '\t',
check.names = T,
row.names = 1
);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(
as.numeric(as.matrix(data)),
nrow = nrow(data),
dimnames = dimnames
);
colnames(data) <- gsub('[.]', '-', colnames(data));
# 去除低表达基因
data <- data[rowMeans(data)>1, ];
将data分成肿瘤组和正常组,方法同生信课01,将列名按-切分,第4个值如果是01-09就是癌症,10-19是正常,20-29是癌旁(也算正常组)。因此我们只需取出01-09的0,10-19的1
# group:标识每个样本是正常还是癌症
group <- sapply(strsplit(colnames(data), '\\-'), "[", 4); # 取出第4个值,以数组形式返回
# 等效于group <- sapply(strsplit(colnames(data), '\\-'), function(x){x[4]});
group <- sapply(strsplit(group, ''), "[", 1); # 再取出第一个值
group <- gsub("2", "1", group);
# 获取正常组和肿瘤组样本数
con_num <- length(group[group==1]); # 正常组
treat_num <- length(group[group==0]); # 肿瘤组
# 根据肿瘤组和正常组排序,将正常组提前
data1 <- data[, group==1];
data2 <- data[, group==0];
data <- cbind(data1, data2);
# 获取新分组信息type,正常组为1, 肿瘤组为2
type <- c(rep(1, con_num), rep(2, treat_num));
差异分析:
-
首先依次对每行(每个基因)进行分析
-
使用
wilcox.test分析表达量与样本类别的关系,得到p值 -
分别计算正常组和肿瘤组表达量平均值,并取log2,它们的差为
logFC -
分别计算正常组和肿瘤组表达量中位数,它们的差为
diffMed -
当
logFC和diffMed有相同的趋势(都>0或都<0)时,将该基因的分析结果保存到结果矩阵中 -
结果矩阵:列名是基因名、两组表达量平均值、logFC、p值
-
-
根据p值,使用
p.adjust函数获取fdr值,即修正后的p值 -
设定logFC和fdr的阈值,筛选出差异基因
# 对每行(每个基因)进行分析
outTab <- data.frame(); # 结果矩阵
for (i in row.names(data)) {
rt <- data.frame( # 每个基因的表达矩阵
expression=data[i, ], # 某样本的表达量
type=type # 该样本属于哪组
);
wilcoxTest <- wilcox.test(expression~type, data = rt); # wilcox检验
pvalue <- wilcoxTest$p.value; # p值
con_mean <- mean(data[i, 1:con_num]); # 正常组表达量平均值
treat_mean <- mean(data[i, (con_num+1):ncol(data)]); # 肿瘤组表达量平均值
logFC <- log2(treat_mean)-log2(con_mean); # 取log2后相减
con_med <- median(data[i, 1:con_num]); # 正常组表达量中值
treat_med <- median(data[i, (con_num+1):ncol(data)]); # 肿瘤组表达量中值
diffMed <- treat_med-con_med; # 相减
if((logFC>0&&diffMed>0) || (logFC<0&&diffMed<0)){ # 如果有相同趋势
outTab <- rbind(
outTab,
cbind(
gene = i, # 基因名
conMean = con_mean, # 正常组表达量平均值
treatMean = treat_mean, # 肿瘤组表达量平均值
logFC = logFC, # logFC
pValue = pvalue # p值
)
);
}
}
# 计算fdr值
pvalue <- outTab[, "pValue"];
fdr <- p.adjust(as.numeric(as.vector(pvalue)), method = "fdr");
outTab <- cbind(outTab, fdr=fdr);
# 保存数据--全部基因
write.table(
outTab,
file = "save_data\\TCGA.all.Wilcoxon.txt",
sep = '\t',
row.names = F,
quote = F
);
# 筛选差异基因
logFC_filter <- 1;
fdr_filter <- 0.05;
outDiff <- outTab[
abs(as.numeric(as.vector(outTab$logFC)))>logFC_filter , ];
outDiff <- outDiff[
as.numeric(as.vector(outDiff$fdr))<fdr_filter
, ];
# 保存数据--差异基因
write.table(
outDiff,
file = "save_data\\TCGA.diff.Wilcoxon.txt",
sep = '\t',
row.names = F,
quote = F
);
热图:需要两组数据
-
每个基因在各样本中的表达量(行名是基因名,列名是样本名),表达量+0.01后取log2
-
一个用于标识每个样本属于哪组的vector
gene_num <- 50; # 展示基因的数目
outDiff <- outDiff[order(as.numeric(as.vector(outDiff$logFC))),]; # 按logFC排序
diff_gene <- as.vector(outDiff[, 1]); # 差异基因名称
diff_gene_len <- length(diff_gene); # 差异基因数量
hm_gene <- c(); # 绘制热图的基因
if(diff_gene_len>2*gene_num){ # 如果差异基因数量多于100
hm_gene <- diff_gene[c(1:gene_num, (diff_gene_len-gene_num+1):diff_gene_len)]; # 取前50个和后50个
} else{
hm_gene <- diff_gene; # 否则全取
}
hm_exp <- log2(data[hm_gene, ]+0.01); # 表达量,加0.01后取log2(避免0值)
type <- c(rep("Normal", con_num), rep("Tumor", treat_num)); # 标识组别
names(type) <- colnames(data); # 建立组别与样本名的对照
type <- as.data.frame(type); # 转为df
pdf( # 创建画图文件
file = "save_data\\heatmap.pdf",
width = 10,
height = 6.5
);
pheatmap( # 开始画图
hm_exp,
annotation = type,
color = colorRampPalette(c(rep('blue', 5),"white", rep("red", 5)))(50),
cluster_cols = F,
show_colnames = F,
scale = "row",
fontsize = 8,
fontsize_row = 5,
fontsize_col = 8
);
dev.off(); # 关闭文件
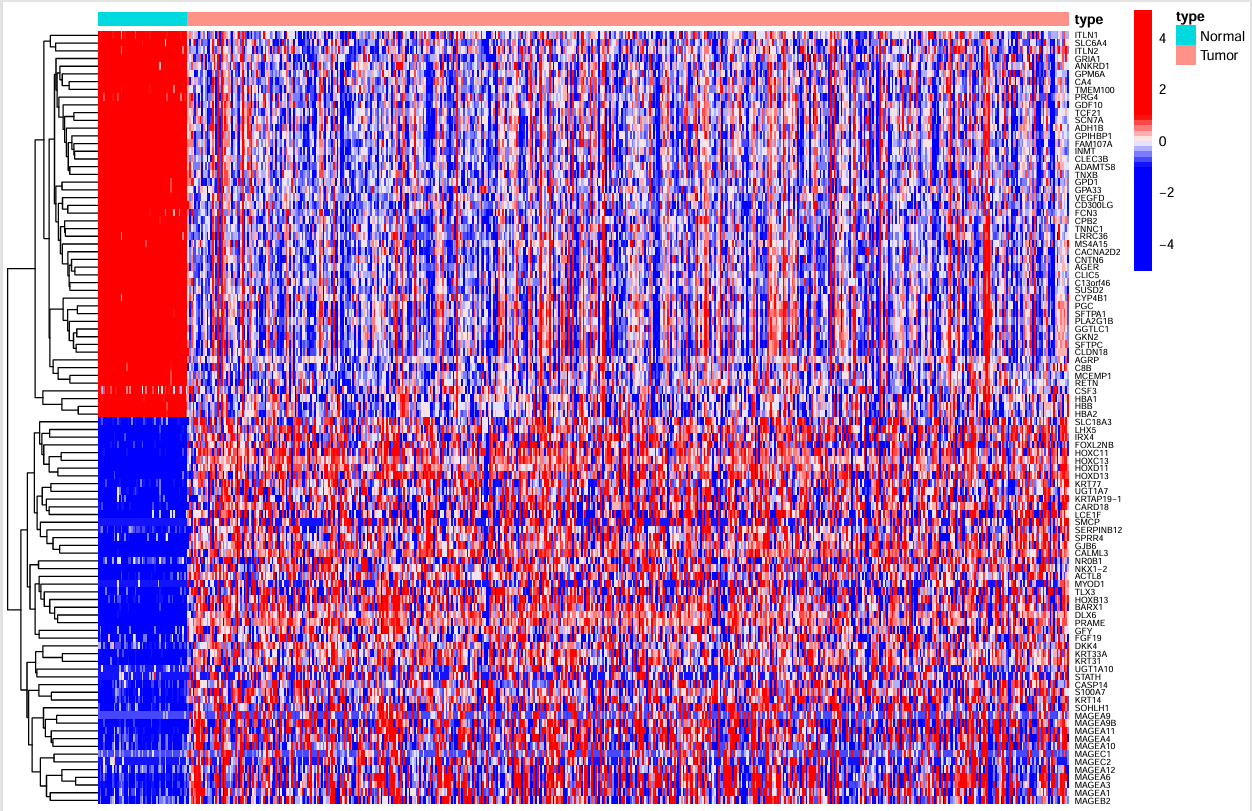
左面的树状线是基因聚类,右面(纵轴)是基因名称,横轴为不同的样本,每个点的颜色表示表达量大小,最上面的type标识每个样本是肿瘤/正常组
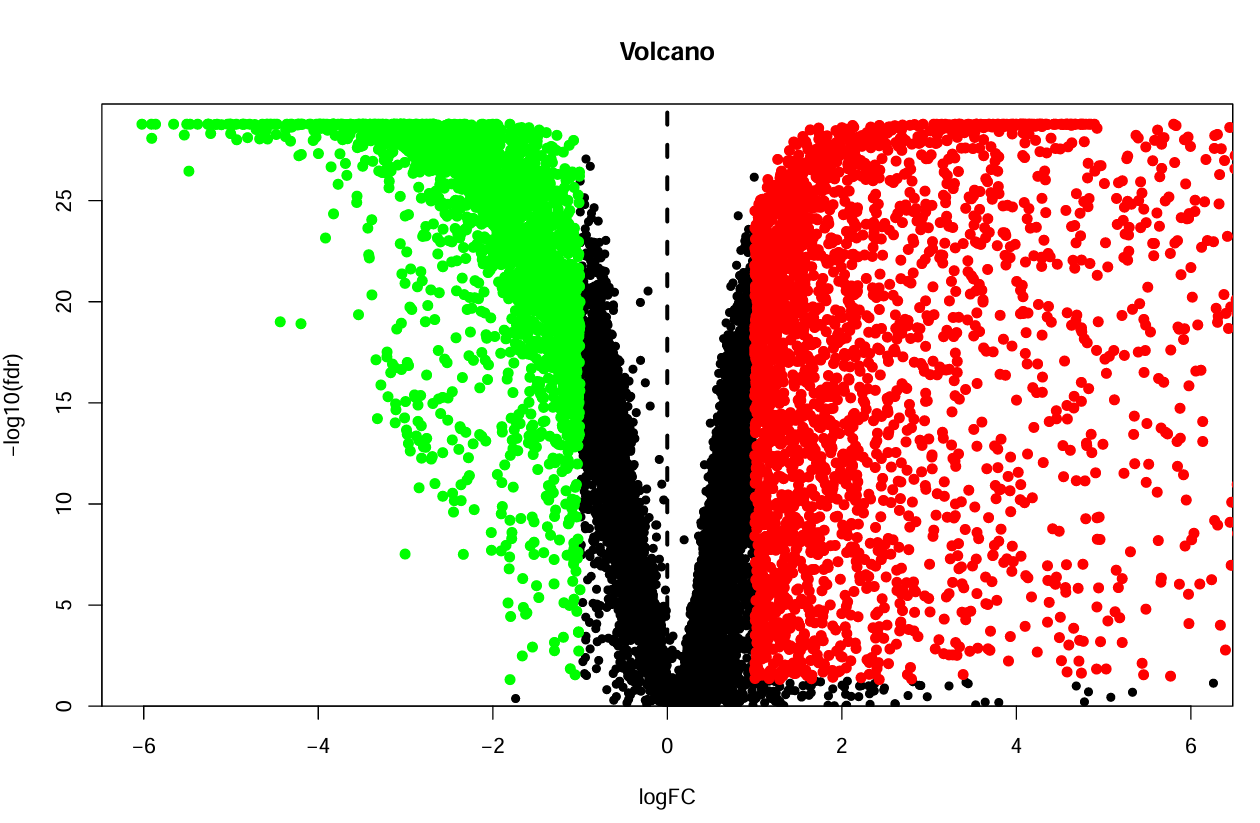
火山图:
横坐标是logFC,纵坐标是-log10(fdr),先画出所有的点,之后以x=±logFC_filter为分界线,左半部分点为绿色,右半部分点为红色
x_max <- 6;
y_max <- max(-log10(outTab$fdr))+1; # xy轴极值
pdf( # 创建画图文件
file = "save_data\\vol.pdf",
width = 10,
height = 6.5
);
# 所有的点
plot(
as.numeric(as.vector(outTab$logFC)),
-log10(outTab$fdr), # 横纵坐标
xlab = "logFC",
ylab = "-log10(fdr)", # xy轴标签
main = "Volcano", # 标题
ylim = c(0, y_max),
xlim = c(-x_max, x_max), # xy轴范围
yaxs = "i", # 设置坐标轴范围与给定作图范围完全相同
pch = 20, # 点的形状
cex = 1.2 # 绘图字符和符号相对于默认大小的缩放比例
);
# x=logFC_filter右边的点,需要对数据集再次进行筛选
diffSub1 <- subset(outTab, fdr<fdr_filter);
diffSub1 <- subset(diffSub1, as.numeric(as.vector(logFC))>logFC_filter);
points(
as.numeric(as.vector(diffSub1$logFC)),
-log10(diffSub1$fdr),
pch = 20,
col = "red",
cex = 1.5
);
# x=-logFC_filter左边的点,也需要对数据集再次进行筛选
diffSub2 <- subset(outTab, fdr<fdr_filter);
diffSub2 <- subset(diffSub2, as.numeric(as.vector(logFC))<(-logFC_filter));
points(
as.numeric(as.vector(diffSub2$logFC)),
-log10(diffSub2$fdr),
pch = 20,
col = "green",
cex = 1.5
);
# x=0的虚线
abline(v=0, lty=2, lwd=3);
dev.off();
count值
limma
需要包:limma、pheatmap、edgeR
if(!require("edgeR", quietly = T))
{
library("BiocManager");
BiocManager::install("edgeR");
library("edgeR");
}
library("pheatmap");
library("limma");
library("tidyverse");
读取文件并分组:
data <- read.table(
"save_data\\TCGA_LUSC_count.txt",
header = T,
sep = '\t',
check.names = T,
row.names = 1
);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(
as.numeric(as.matrix(data)),
nrow = nrow(data),
dimnames = dimnames
);
colnames(data) <- gsub('[.]', '-', colnames(data));
data <- data[rowMeans(data)>1, ];
group <- sapply(strsplit(colnames(data), '\\-'), "[", 4);
group <- sapply(strsplit(group, ''), "[", 1);
group <- gsub("2", "1", group);
con_num <- length(group[group==1]);
treat_num <- length(group[group==0]);
data1 <- data[, group==1];
data2 <- data[, group==0];
data <- cbind(data1, data2);
type <- c(rep(1, con_num), rep(2, treat_num));
type <- factor(type);
design <- model.matrix(~0+type);
rownames(design) <- colnames(data);
进行差异分析:
DGElist <- DGEList(counts=data, group=type);
keep_gene_index <- rowSums(cpm(DGElist)>1) >= 2;
DGElist <- DGElist[keep_gene_index, , keep.lib.sizes=F];
DGElist <- calcNormFactors(DGElist);
v <- voom(DGElist, design, plot = T, normalize="quantile");
fit <- lmFit(v, design);
colnames(design) <- c("normal", "tumor");
cont.matrix <- makeContrasts(contrasts = c('tumor-normal'), levels = design);
fit2 <- contrasts.fit(fit, cont.matrix);
fit2 <- eBayes(fit2);
nrDEG_limma_voom <- topTable(fit2, coef = 'tumor-normal', n = Inf);
nrDEG_limma_voom <- na.omit(nrDEG_limma_voom);
筛选:
padj <- 0.05;
logFC <- 1;
outDiff <- nrDEG_limma_voom[nrDEG_limma_voom$adj.P.Val<padj, ];
outDiff <- rbind(
outDiff[outDiff$logFC>logFC, ],
outDiff[outDiff$logFC<(-logFC), ]
);
write.table(
data.frame(
ID = rownames(outDiff),
outDiff
),
file = "save_data\\TCGA.diff.limma.txt",
sep = '\t',
row.names = F,
quote = F
);
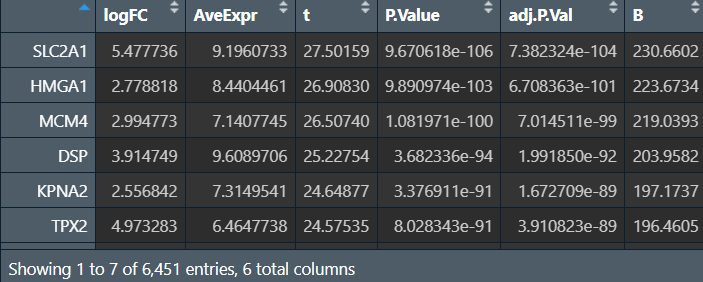
之后用outDiff画图的过程同前,下同
edgeR
需要包:limma、pheatmap、edgeR
library("edgeR");
library("pheatmap");
library("limma");
library("tidyverse");
读取文件并分组:
data <- read.table(
"save_data\\TCGA_LUSC_count.txt",
header = T,
sep = '\t',
check.names = T,
row.names = 1
);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(
as.numeric(as.matrix(data)),
nrow = nrow(data),
dimnames = dimnames
);
colnames(data) <- gsub('[.]', '-', colnames(data));
data <- data[rowMeans(data)>1, ];
group <- sapply(strsplit(colnames(data), '\\-'), "[", 4);
group <- sapply(strsplit(group, ''), "[", 1);
group <- gsub("2", "1", group);
con_num <- length(group[group==1]);
treat_num <- length(group[group==0]);
data1 <- data[, group==1];
data2 <- data[, group==0];
data <- cbind(data1, data2);
type <- c(rep(1, con_num), rep(2, treat_num));
type <- factor(type);
design <- model.matrix(~0+type);
rownames(design) <- colnames(data);
差异分析:
DGElist <- DGEList(counts=data, group=type);
keep_gene_index <- rowSums(cpm(DGElist)>1) >= 2;
DGElist <- DGElist[keep_gene_index, , keep.lib.sizes=F];
DGElist <- calcNormFactors(DGElist);
DGElist <- estimateGLMCommonDisp(DGElist, design);
DGElist <- estimateGLMTrendedDisp(DGElist, design);
DGElist <- estimateGLMTagwiseDisp(DGElist, design);
fit <- glmFit(DGElist, design);
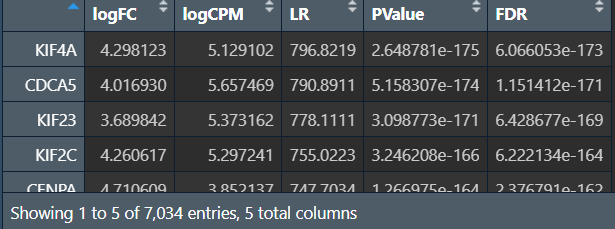
results <- glmLRT(fit, contrast = c(-1, 1));
nrDEG_edgeR <- topTags(results, n = nrow(DGElist));
nrDEG_edgeR <- as.data.frame(nrDEG_edgeR);
筛选(同前):
padj <- 0.05;
logFC <- 1;
outDiff <- nrDEG_edgeR[nrDEG_edgeR$FDR<padj, ];
outDiff <- rbind(
outDiff[outDiff$logFC>logFC, ],
outDiff[outDiff$logFC<(-logFC), ]
);
write.table(
data.frame(
ID = rownames(outDiff),
outDiff
),
file = "save_data\\TCGA.diff.edgeR.txt",
sep = '\t',
row.names = F,
quote = F
);
DESeq2
需要包:DESeq2
与生信课01中的类似,只是这里没有分成训练组和验证组
if(!require("DESeq2", quietly = T))
{
library("BiocManager");
BiocManager::install("DESeq2");
library("DESeq2");
}
读取数据并分组:
data <- read.table(
"save_data\\TCGA_LUSC_count.txt",
header = T,
sep = '\t',
check.names = T,
row.names = 1
);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(
as.numeric(as.matrix(data)),
nrow = nrow(data),
dimnames = dimnames
);
colnames(data) <- gsub('[.]', '-', colnames(data));
data <- data[rowMeans(data)>1, ];
group <- sapply(strsplit(colnames(data), '\\-'), "[", 4);
group <- sapply(strsplit(group, ''), "[", 1);
group <- gsub("2", "1", group);
con_num <- length(group[group==1]);
treat_num <- length(group[group==0]);
data1 <- data[, group==1];
data2 <- data[, group==0];
data <- cbind(data1, data2);
type <- c(rep(1, con_num), rep(2, treat_num));
condition <- factor(type);
coldata <- data.frame(row.names = colnames(data), condition);
差异分析:
dds <- DESeqDataSetFromMatrix(
countData = data,
colData = coldata,
design = ~condition
);
dds$condition <- relevel(dds$condition, ref = "1"); # 指定为1的行是正常组(对照组)
dds <- DESeq(dds);
allDEG2 <- as.data.frame(results(dds));
筛选(同前):
padj <- 0.05;
logFC <- 1;
outDiff <- allDEG2[allDEG2$padj<padj, ];
outDiff <- rbind(
outDiff[outDiff$log2FoldChange>logFC, ],
outDiff[outDiff$log2FoldChange<(-logFC), ]
);
write.table(
data.frame(
ID = rownames(outDiff),
outDiff
),
file = "save_data\\TCGA.diff.DESeq2.txt",
sep = '\t',
row.names = F,
quote = F
);
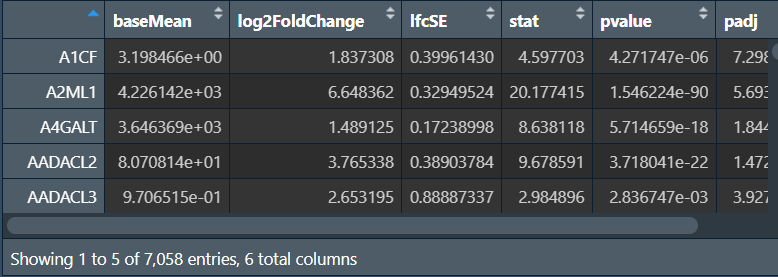
查看以上4种分析结果
使用包:VennDiagram
if(!require("VennDiagram", quietly = T))
{
install.packages("VennDiagram");
library("VennDiagram");
}
载入4组数据,获取4种方法得到差异基因名
data_name <- c("edgeR", "limma", "DESeq2", "Wilcoxon");
file_path <- c("save_data\\TCGA.diff.edgeR.txt", "save_data\\TCGA.diff.limma.txt", "save_data\\TCGA.diff.DESeq2.txt", "save_data\\TCGA.diff.Wilcoxon.txt");
data_list <- list(); # 结果列表
for (i in 1:length(file_path)) {
data <- read.table(file_path[i], header = T, sep = '\t', check.names = F, row.names = 1);
data_list[[data_name[i]]] <- rownames(data);
}
画图:
venn.diagram(
x = data_list,
filename = "save_data\\VN.png",
fill = c("dodgerblue", "goldenrod1", "darkorange1", "green")
);
单因素cox回归
使用包survival、survminer
if(!require("survival", quietly = T))
{
install.packages("survival");
library("survival");
}
if(!require("survminer", quietly = T))
{
install.packages("survminer");
library("survminer");
}
读取文件:
-
表达矩阵
TCGA_LUSC_TPM.txt -
差异基因
TCGA.diff.limma.txt -
生存状况
clinical.xlsx
# 表达矩阵
tpm <- read.table("save_data\\TCGA_LUSC_TPM.txt", check.names = F, row.names = 1, sep = '\t', header = T);
dimnames <- list(rownames(tpm), colnames(tpm));
tpm <- matrix(as.numeric(as.matrix(tpm)), nrow = nrow(tpm), dimnames = dimnames);
# 差异基因
limma <- read.table("save_data\\TCGA.diff.limma.txt", check.names = F, row.names = 1, sep = '\t', header = T);
tpm <- tpm[rownames(limma), ]; # 获得差异基因的表达矩阵
tpm <- t(tpm); # 转置,使行名为样本名,格式与生存信息相同
rownames(tpm) <- substr(rownames(tpm), 1, 12); # 样本名仅保留前12个字符,格式与生存信息相同
# 生存信息
library("readxl");
cli <- read_excel("save_data\\clinical.xlsx");
library("tidyverse");
cli <- column_to_rownames(cli, "bcr_patient_barcode"); # 样本名为行名
cli <- cli[, c("survival_time", "vital_status")]; # 只保留生存时间、生存状态列
colnames(cli) <- c("time", "state"); # 改列名
cli$time <- as.numeric(cli$time); # 改列类型
cli <- cli[cli$time>=30, ]; # 只保留生存时间>=30的行
cli <- cli[!is.na(cli$time), ];
cli <- cli[!is.na(cli$state), ]; # 过滤掉NA
cli$time <- cli$time/365; # 时间以年为单位
cli$state <- ifelse(cli$state=='Alive', 0, 1); # 死亡用1表示,存活用0表示
library("writexl");
write_xlsx(data.frame(ID = rownames(cli), cli), "save_data\\time_LUSC.xlsx");
将生存信息与表达矩阵进行合并,提取共含的样本
same_sample <- intersect(rownames(tpm), rownames(cli)); # 共同样本名
tpm <- tpm[same_sample, ];
cli <- cli[same_sample, ]; # 过滤
rt <- cbind(cli, tpm); # 合并
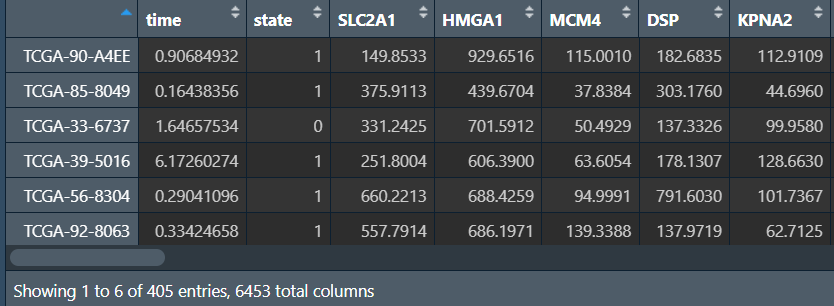
cox回归:
p.value <- 0.01; # p值的阈值
outTab <- data.frame(); # 结果矩阵
for (i in colnames(rt[, 3:ncol(rt)])) { # i是基因名
cox <- coxph(Surv(time, state) ~ rt[, i], data = rt); # 分析生存信息与某个基因表达量的关系
cox_summary <- summary(cox); # 对结果进行分析
cox_p <- cox_summary$coefficients[, "Pr(>|z|)"]; # p值
if(cox_p<p.value){ # 根据p值进行筛选
outTab <- rbind( # 将结果添加到结果矩阵中
outTab,
cbind(
id = i, # 基因名
HR = cox_summary$conf.int[, "exp(coef)"], # 风险比
HR.95L = cox_summary$conf.int[, "lower .95"], # 95CI下限
HR.95H = cox_summary$conf.int[, "upper .95"], # 95CI上限
pvalue = cox_p # p值
)
);
}
}
# 保存结果
write.table(
outTab,
file = "save_data\\uniCox.txt",
row.names = F, sep = '\t', quote = F
);
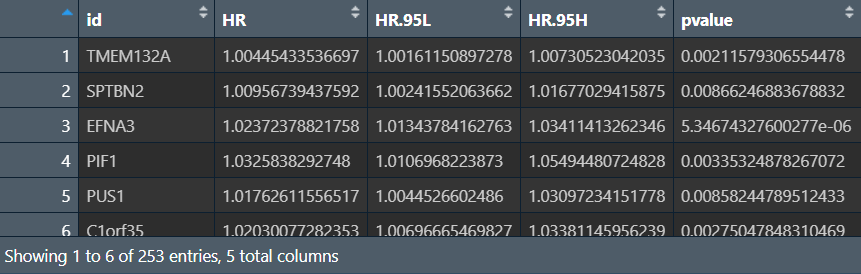
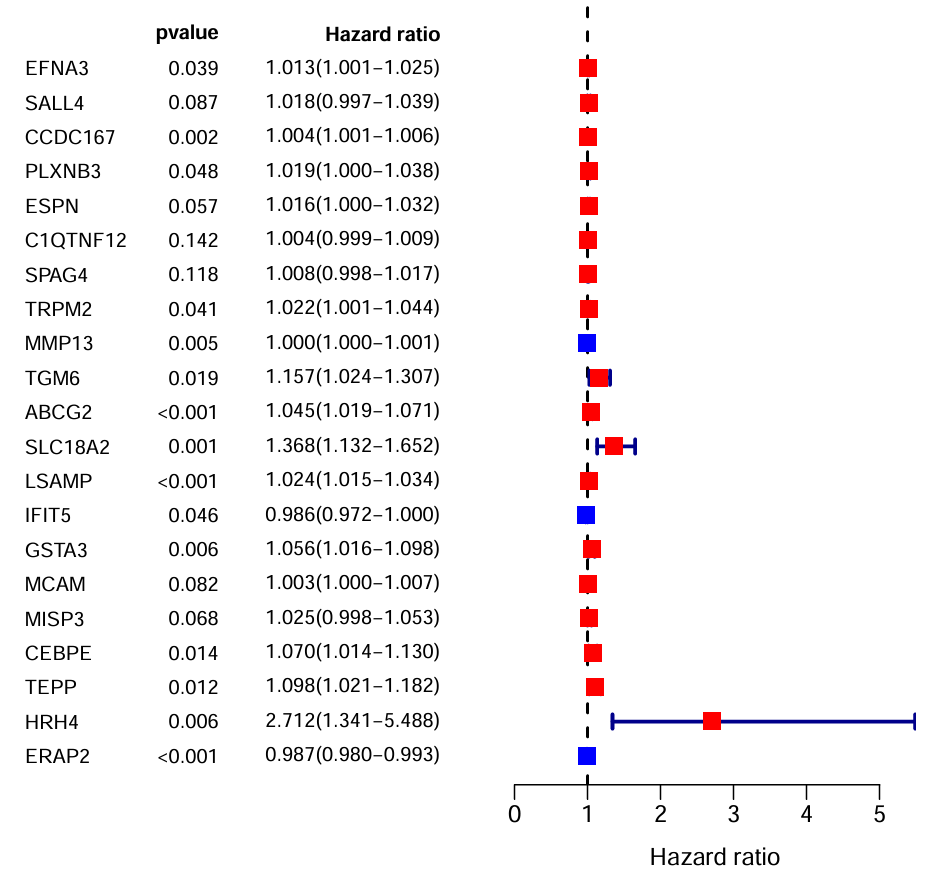
画图的数据准备:基因名、HR、p值
rt <- read.table( "save_data\\uniCox.txt", check.names = F, row.names = 1, sep = '\t', header = T);
show_num <- 20; # 展示基因的数量
rt <- rt[sample(1:nrow(rt), show_num, replace = F), ]; # 为方便展示,这里只画出前20个基因
gene <- rownames(rt); # 基因名
hr <- sprintf("%.3f", rt$HR); # 风险比,sprintf函数用法同C中的printf,这里是保留3位小数
hrLow <- sprintf("%.3f", rt$HR.95L); # 95CI下限
hrHigh <- sprintf("%.3f", rt$HR.95H); # 95CI上限
Hazard.ratio <- paste0(hr, "(", hrLow, "-", hrHigh, ")"); # 风险比HR(包括95CI)
pVal <- ifelse(
rt$pvalue<0.001,
"<0.001",
sprintf("%.3f", rt$pvalue)
);
画图:使用layout函数进行多图布局,接收一个矩阵,表示每个格子对应的图,比设置par更方便,更多关于layout函数
n <- nrow(rt);
nRow <- n+1;
ylim <- c(1, nRow); # y轴上下限
layout_matrix <- matrix(c(1, 2), nc=2);
layout_matrix:
[,1] [,2]
[1,] 1 2
表示左右分别有一个图,左边是文字,右边是图主体
pdf(
file = "save_data\\uniCoxforest.pdf",
width = 7,
height = nrow(rt)/13+5
);
layout(layout_matrix, width = c(3, 2.5));
# 左侧
xlim <- c(0, 3); # x轴上下限
par(mar=c(4, 2.5, 2, 1)); # 设置图形边距
plot(
1,
xlim = xlim,
ylim = ylim,
type = "n", # 不画图,只写文字
axes = F, # 无坐标轴
xlab = "",
ylab = "" # 无标题
);
text.cex <- 0.8; # 文字大小(缩放比例)
text(0, n:1, gene, adj=0, cex=text.cex); # 基因名 adj指定对齐方式:0-左对齐 1-右对齐
text(1.4, n:1, pVal, adj=1, cex=text.cex); # p值列表
text(1.4, n+1, 'pvalue', adj=1, cex=text.cex, font=2); # p-value标题
text(3, n:1, Hazard.ratio, adj=1, cex=text.cex); # HR值列表
text(3, n+1, 'Hazard ratio', adj=1, cex=text.cex, font=2); # Hazard ratio标题
# 右侧
xlim <- c(0, max(as.numeric(hrLow), as.numeric(hrHigh))); # x轴上下限
par(mar=c(4, 1, 2, 1), mgp=c(2, 0.5, 0)); # 设定图形边距,标题、坐标轴名称、坐标轴距图形边框的距离
plot(
1,
xlim = xlim,
ylim = ylim,
type = "n", # 不画图,只写文字
axes = F, # 无坐标轴
xlab = "Hazard ratio",
ylab = "",
xaxs = "i" # 指定x轴起点
);
abline(v=1, col="black", lty=2, lwd=2); # x=1处的虚线
arrows( # 标识HR的CI95区间,即图中方块点两侧的T型线
as.numeric(hrLow),
n:1,
as.numeric(hrHigh),
n:1,
angle = 90,
code = 3,
length = 0.05,
col = "darkblue",
lwd = 2.5
);
boxcolor <- ifelse(as.numeric(hr)>1, "red", "blue"); # HR大于1红色,小于1蓝色
points( # 方块型的点
as.numeric(hr),
n:1,
pch = 15,
col = boxcolor,
cex = 1.6
);
axis(1); # 在图形的下边绘制坐标轴
dev.off();
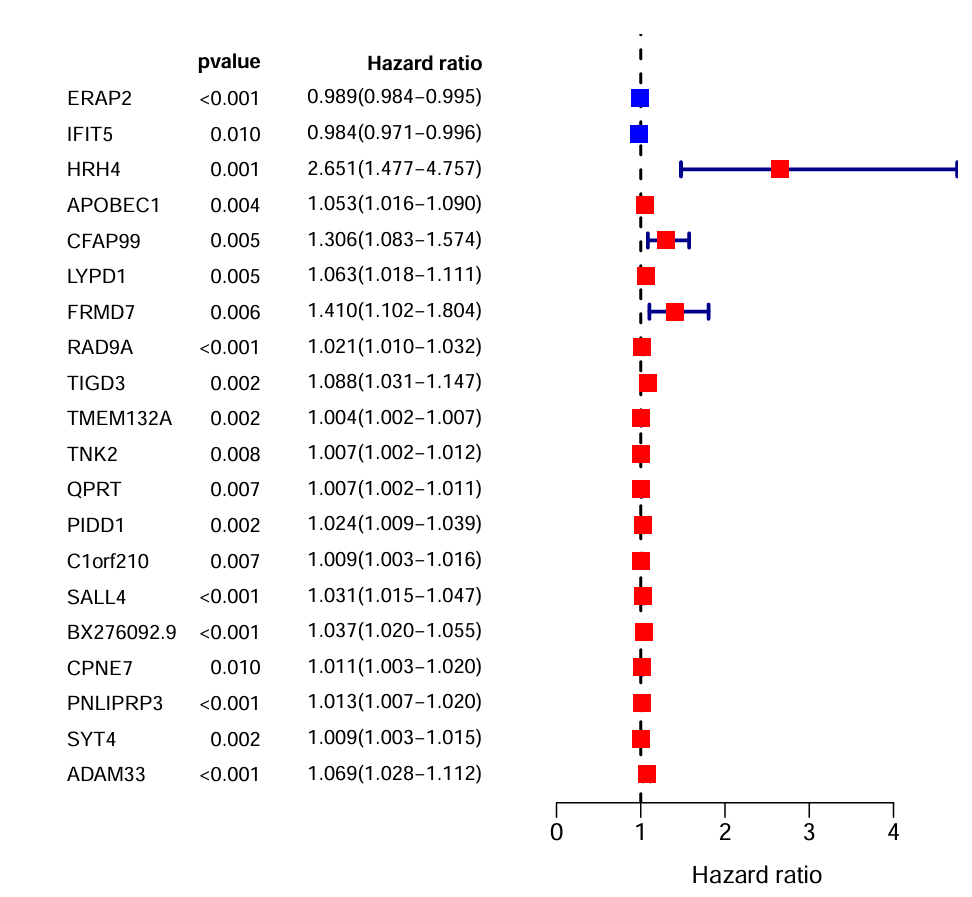
注:画图函数的顺序决定了结果图中谁覆盖谁,这里我们想让方块点>T型线>x=1虚线
代码汇总:
rm(list=ls());
if(!require("survival", quietly = T))
{
install.packages("survival");
library("survival");
}
if(!require("survminer", quietly = T))
{
install.packages("survminer");
library("survminer");
}
tpm <- read.table("save_data\\TCGA_LUSC_TPM.txt", check.names = F, row.names = 1, sep = '\t', header = T);
dimnames <- list(rownames(tpm), colnames(tpm));
tpm <- matrix(as.numeric(as.matrix(tpm)), nrow = nrow(tpm), dimnames = dimnames);
limma <- read.table("save_data\\TCGA.diff.limma.txt", check.names = F, row.names = 1, sep = '\t', header = T);
tpm <- tpm[rownames(limma), ];
tpm <- t(tpm);
rownames(tpm) <- substr(rownames(tpm), 1, 12);
library("readxl");
cli <- read_excel("save_data\\clinical.xlsx");
library("tidyverse");
cli <- column_to_rownames(cli, "bcr_patient_barcode");
cli <- cli[, c("survival_time", "vital_status")];
colnames(cli) <- c("time", "state");
cli$time <- as.numeric(cli$time);
cli <- cli[cli$time>=30, ];
cli <- cli[!is.na(cli$time), ];
cli <- cli[!is.na(cli$state), ];
cli$time <- cli$time/365;
cli$state <- ifelse(cli$state=='Alive', 0, 1);
same_sample <- intersect(rownames(tpm), rownames(cli));
tpm <- tpm[same_sample, ];
cli <- cli[same_sample, ];
rt <- cbind(cli, tpm);
p.value <- 0.01;
outTab <- data.frame();
for (i in colnames(rt[, 3:ncol(rt)])) {
cox <- coxph(Surv(time, state) ~ rt[, i], data = rt);
cox_summary <- summary(cox);
cox_p <- cox_summary$coefficients[, "Pr(>|z|)"];
if(cox_p<p.value){
outTab <- rbind(outTab, cbind(id = i, HR = cox_summary$conf.int[, "exp(coef)"], HR.95L = cox_summary$conf.int[, "lower .95"], HR.95H = cox_summary$conf.int[, "upper .95"], pvalue = cox_p));}}
write.table(outTab, file = "save_data\\uniCox.txt", row.names = F, sep = '\t', quote = F);
rm(list=ls());
rt <- read.table( "save_data\\uniCox.txt", check.names = F, row.names = 1, sep = '\t', header = T);
show_num <- 20;
rt <- rt[sample(1:nrow(rt), show_num, replace = F), ];
gene <- rownames(rt);
hr <- sprintf("%.3f", rt$HR);
hrLow <- sprintf("%.3f", rt$HR.95L);
hrHigh <- sprintf("%.3f", rt$HR.95H);
Hazard.ratio <- paste0(hr, "(", hrLow, "-", hrHigh, ")");
pVal <- ifelse(rt$pvalue<0.001, "<0.001", sprintf("%.3f", rt$pvalue));
n <- nrow(rt);
nRow <- n+1;
ylim <- c(1, nRow);
layout_matrix <- matrix(c(1, 2), nc=2);
pdf(file = "save_data\\uniCoxforest.pdf", width = 7, height = nrow(rt)/13+5);
layout(layout_matrix, width = c(3, 2.5));
xlim <- c(0, 3);
par(mar=c(4, 2.5, 2, 1));
plot(1, xlim = xlim, ylim = ylim, type = "n", axes = F, xlab = "", ylab = "" );
text.cex <- 0.8;
text(0, n:1, gene, adj=0, cex=text.cex);
text(1.4, n:1, pVal, adj=1, cex=text.cex);
text(1.4, n+1, 'pvalue', adj=1, cex=text.cex, font=2);
text(3, n:1, Hazard.ratio, adj=1, cex=text.cex);
text(3, n+1, 'Hazard ratio', adj=1, cex=text.cex, font=2);
xlim <- c(0, max(as.numeric(hrLow), as.numeric(hrHigh)));
par(mar=c(4, 1, 2, 1), mgp=c(2, 0.5, 0));
plot(1, xlim = xlim, ylim = ylim, type = "n", axes = F, xlab = "Hazard ratio", ylab = "", xaxs = "i");
abline(v=1, col="black", lty=2, lwd=2);
arrows(as.numeric(hrLow), n:1, as.numeric(hrHigh), n:1, angle = 90, code = 3, length = 0.05, col = "darkblue", lwd = 2.5);
boxcolor <- ifelse(as.numeric(hr)>1, "red", "blue");
points(as.numeric(hr), n:1, pch = 15, col = boxcolor, cex = 1.6);
axis(1);
dev.off();
GO富集分析
需要包:ggtree、clusterProfiler、org.Hs.eg.db、enrichplot、GOplot、R.utils
if(!require("ggtree", quietly = T))
{
library("BiocManager");
BiocManager::install("ggtree");
}
if(!require("clusterProfiler", quietly = T))
{
library("BiocManager");
BiocManager::install("clusterProfiler");
}
if(!require("org.Hs.eg.db", quietly = T))
{
library("BiocManager");
BiocManager::install("org.Hs.eg.db");
}
if(!require("enrichplot", quietly = T))
{
library("BiocManager");
BiocManager::install("enrichplot");
}
if(!require("GOplot", quietly = T))
{
install.packages("GOplot");
}
if(!require("R.utils", quietly = T))
{
install.packages("R.utils");
}
library("clusterProfiler");
library("org.Hs.eg.db");
library("ggplot2");
library("stringi");
library("GOplot");
library("enrichplot");
R.utils::setOption("clusterProfiler.download.method", "auto");
准备数据:单因素cox回归得到的基因symbol
input_diff <- read.table("save_data\\uniCox.txt", check.names = F, row.names = 1, sep = '\t', header = T);
input_gene <- rownames(input_diff); # 取基因symbol
input_gene <- unique(as.vector(input_gene)); # 去重
将基因symbol转为基因id:因为这些都是人的基因,使用org.Hs.eg.db库中的数据库org.Hs.egSYMBOL2EG进行转化,其它物种
entrezIDs <- BiocGenerics::mget(input_gene, org.Hs.egSYMBOL2EG, ifnotfound = NA);
entrezIDs <- as.character(entrezIDs);
gene <- entrezIDs[entrezIDs!='NA']; # 去除NA值
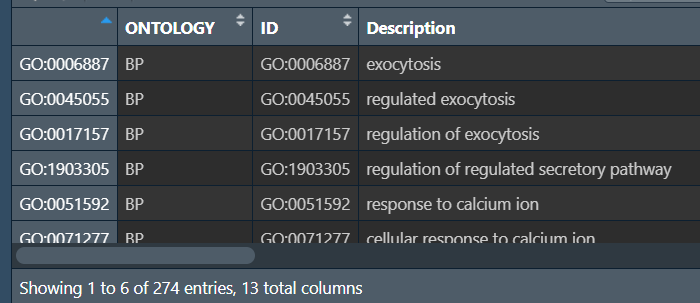
GO富集分析:
# 筛选条件:如果想要结果较多就使用pvalue进行筛选,反之用qvalue
pvalue_filter <- 0.05;
qvalue_filter <- 1;
colorSel <- ifelse(qvalue_filter>0.05, "pvalue", "qvalue");
# GO富集分析(需要联网)
kk <- enrichGO(
gene = gene,
OrgDb = org.Hs.eg.db,
pvalueCutoff = 1,
qvalueCutoff = 1,
ont = "all",
readable = T
);
GO <- as.data.frame(kk);
GO <- GO[GO$pvalue<pvalue_filter, ];
GO <- GO[GO$qvalue<qvalue_filter, ];
# 保存数据
write.table(GO, file = "save_data\\GO.txt", row.names = F, sep = '\t', quote = F)
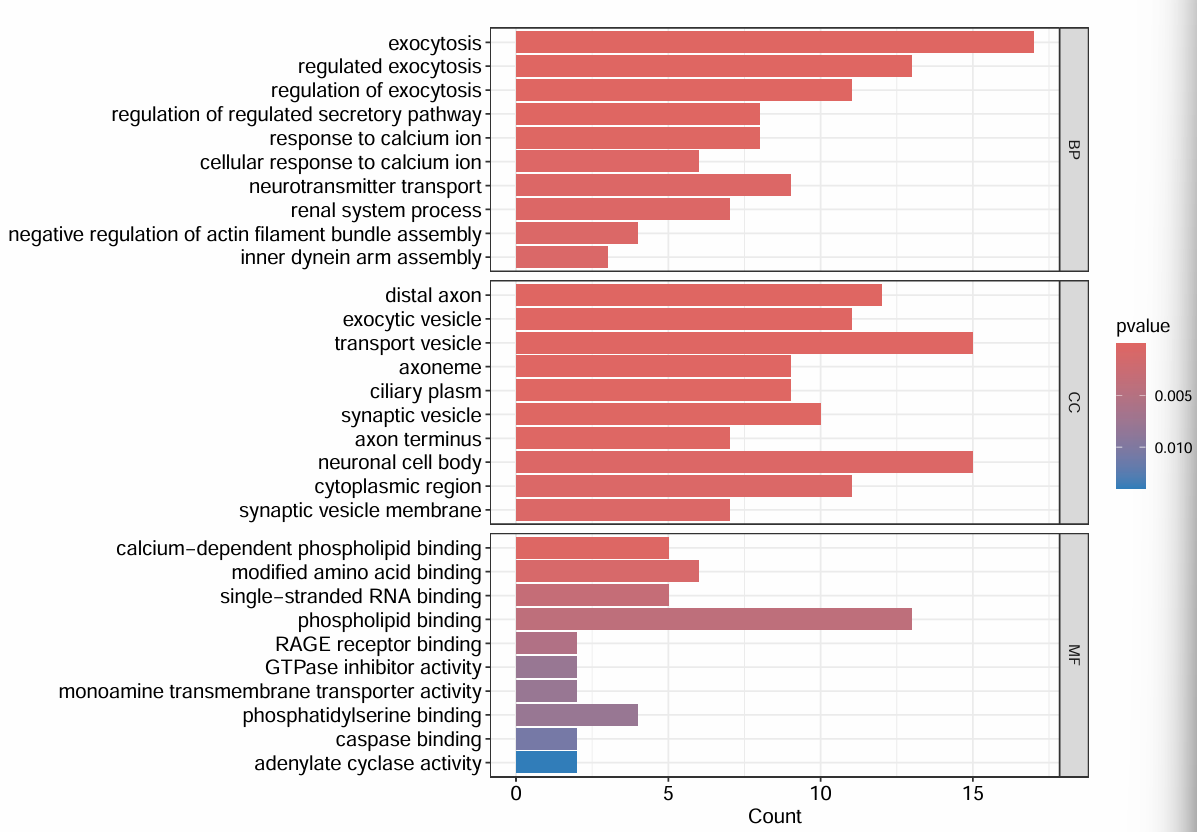
画图:
show_num <- 10; # 只画前10个
# 柱状图
pdf(file = "save_data\\GObarplot.pdf", width = 10, height = 7);
barplot(
kk,
drop = T,
showCategory = show_num,
label_format = 70, # 如果左侧字出现重叠,就增大该值
split = "ONTOLOGY",
color = colorSel
) +
facet_grid(ONTOLOGY~., scale = "free");
dev.off();
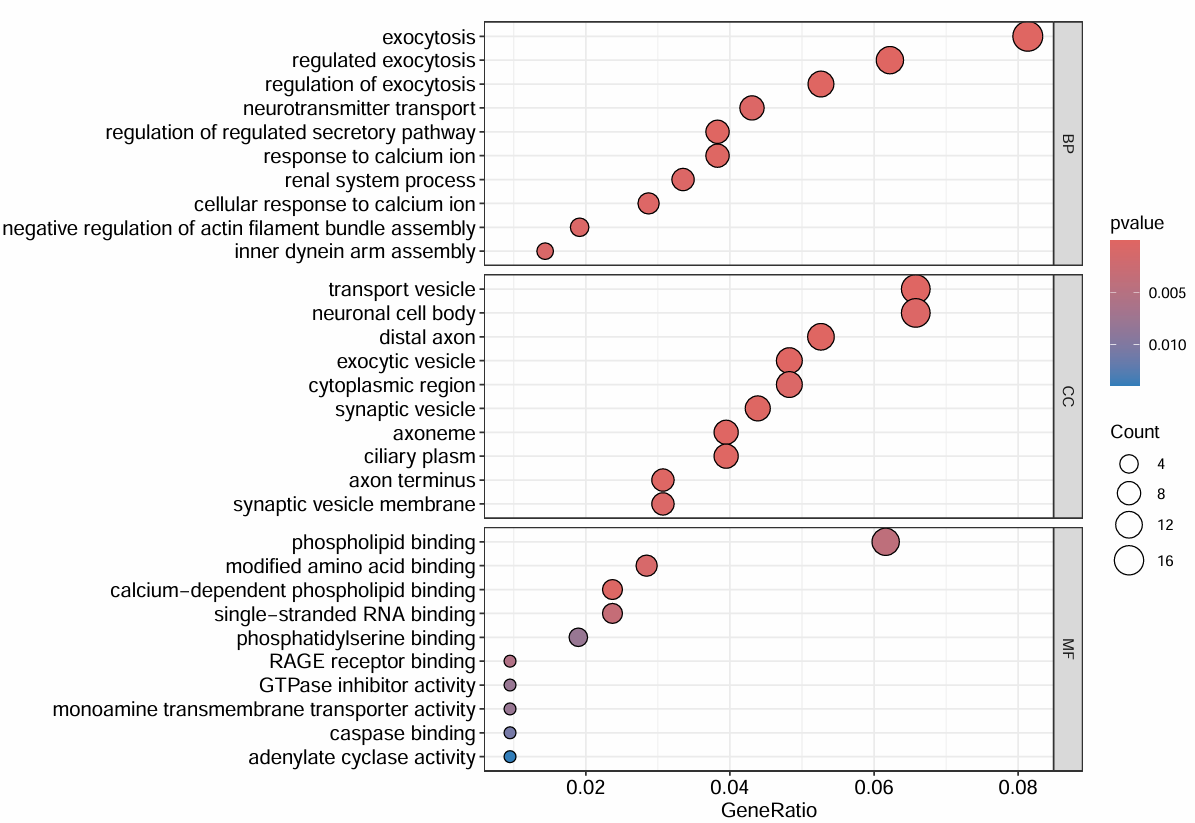
# 气泡图
pdf(file = "save_data\\GObubble.pdf", width = 10, height = 7);
dotplot(
kk,
showCategory = show_num,
orderBy = "GeneRatio",
label_format = 70, # 如果左侧字出现重叠,就增大该值
split = "ONTOLOGY",
color = colorSel
) +
facet_grid(ONTOLOGY~., scale = "free");
dev.off();
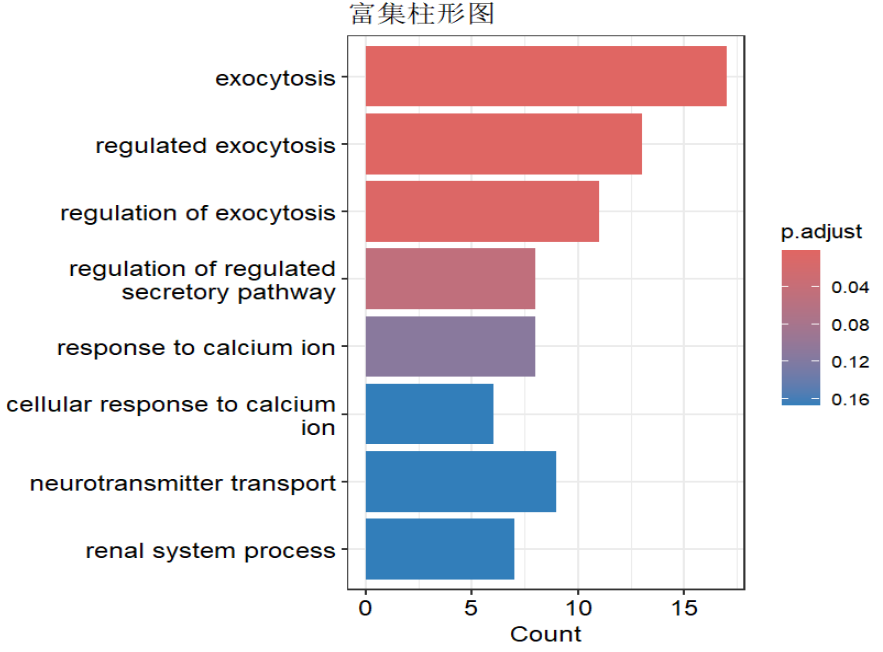
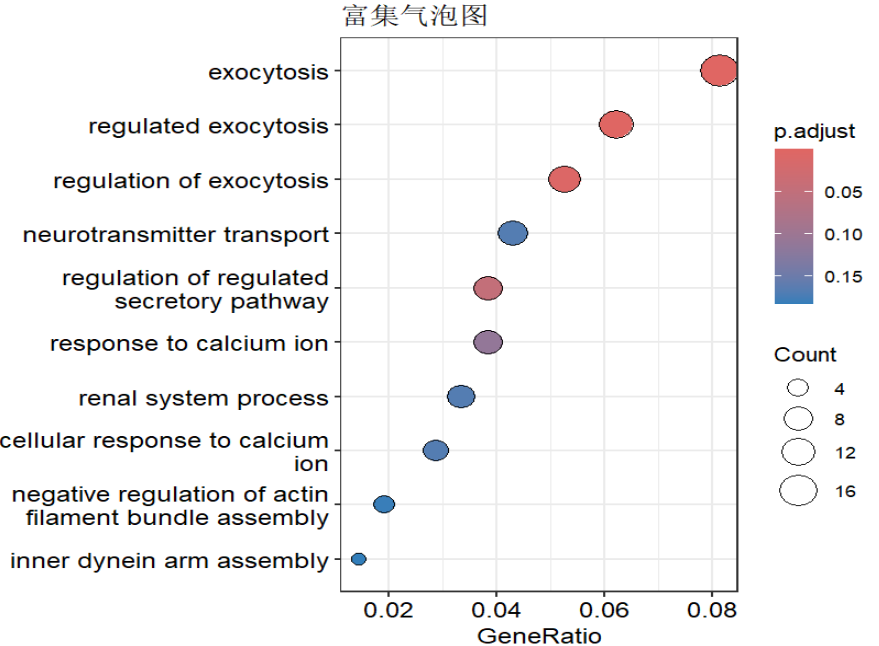
其它clusterProfiler包自带的作图方法:
barplot(kk) + ggtitle("富集柱形图");
dotplot(kk) + ggtitle("富集气泡图");
cnetplot(kk) + ggtitle("网络图1"); # 网络图1:展示富集功能与基因的包含关系
emapplot(pairwise_termsim(kk)) + ggtitle("网络图2"); # 网络图2:展示各富集功能间共有基因关系
heatplot(kk) + ggtitle("热图"); # 热图:展示富集功能与基因的包含关系
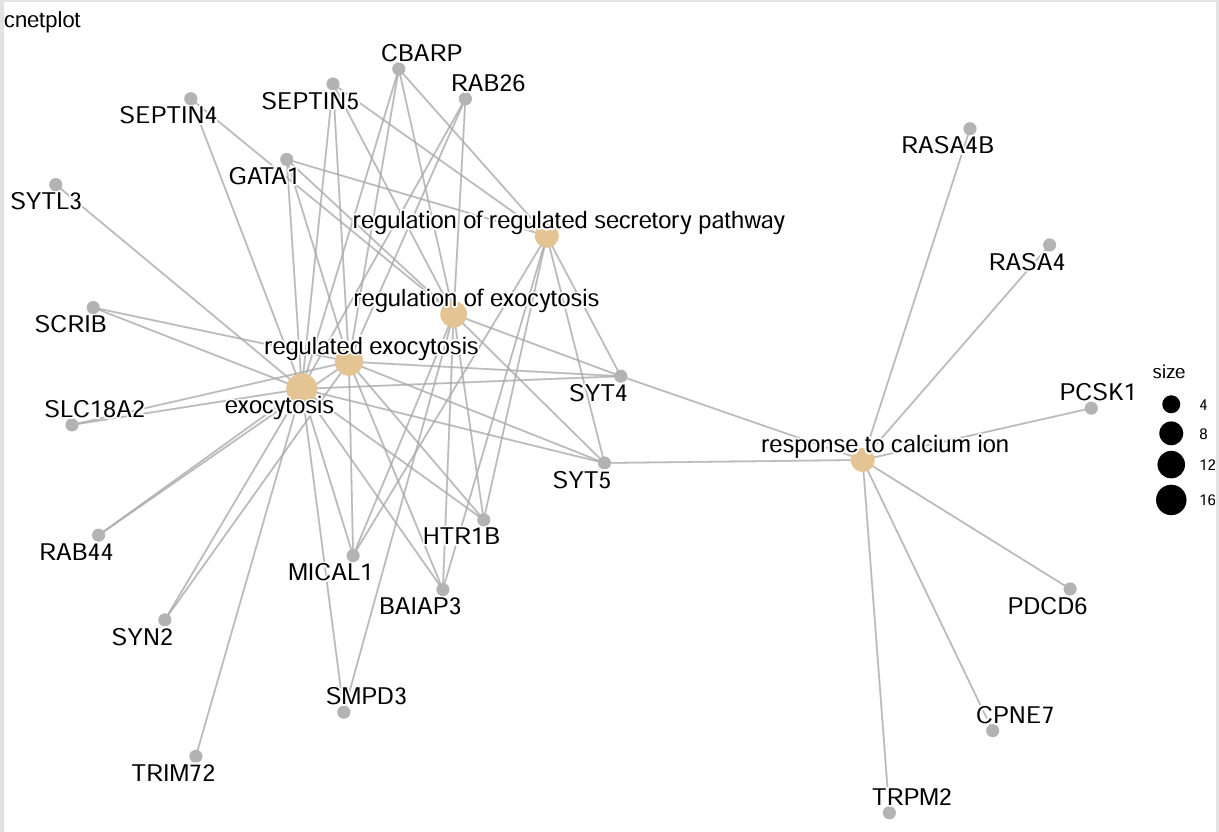
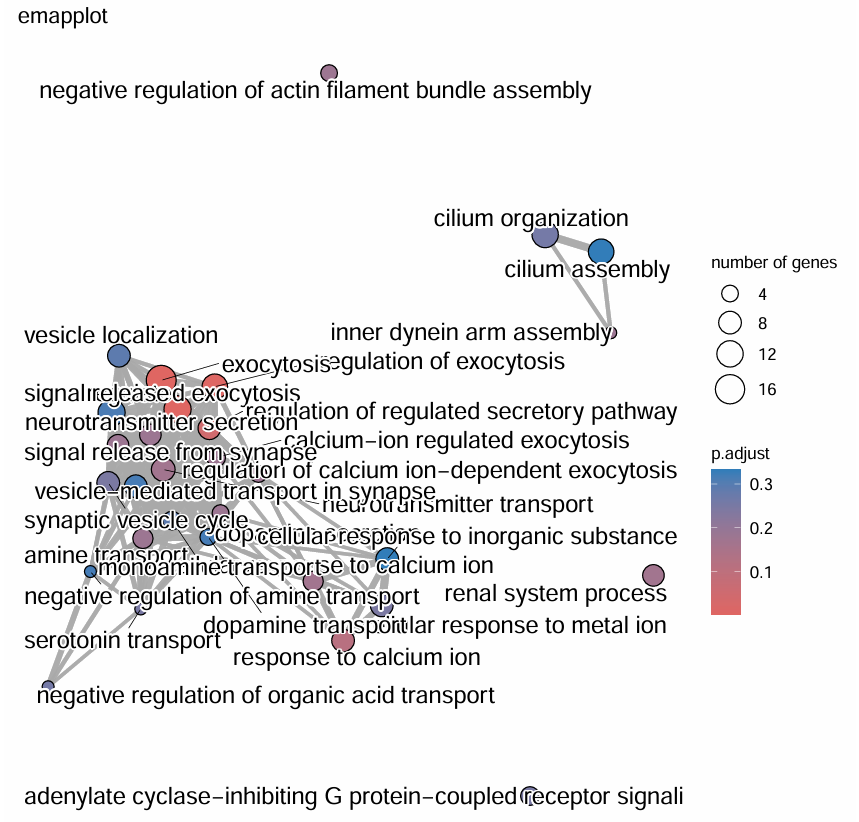

KEGG富集分析
准备数据与GO富集分析相同
library("clusterProfiler");
library("org.Hs.eg.db");
input_diff <- read.table("save_data\\uniCox.txt", check.names = F, row.names = 1, sep = '\t', header = T);
input_gene <- rownames(input_diff);
input_gene <- unique(as.vector(input_gene));
entrezIDs <- BiocGenerics::mget(input_gene, org.Hs.egSYMBOL2EG, ifnotfound = NA);
entrezIDs <- as.character(entrezIDs);
gene <- entrezIDs[entrezIDs!='NA'];
KEGG富集分析:
# 筛选条件:如果想要结果较多就使用pvalue进行筛选
pvalue_filter <- 0.05;
qvalue_filter <- 1;
colorSel <- ifelse(qvalue_filter>0.05, "pvalue", "qvalue");
# GO富集分析(需要联网)
kk <- enrichKEGG(
gene = gene,
organism = "hsa",
pvalueCutoff = 1,
qvalueCutoff = 1,
);
KEGG <- as.data.frame(kk);
KEGG$geneID <- as.character( # 将结果中的基因id转回symbol
sapply(KEGG$geneID, function(x){
paste(
input_gene[
match(strsplit(x, "/")[[1]], entrezIDs)
],
collapse = "/"
);
})
);
KEGG <- KEGG[KEGG$pvalue<pvalue_filter, ];
KEGG <- KEGG[KEGG$qvalue<qvalue_filter, ];
# 保存
write.table(KEGG, file = "save_data\\KEGG.txt", row.names = F, sep = '\t', quote = F);
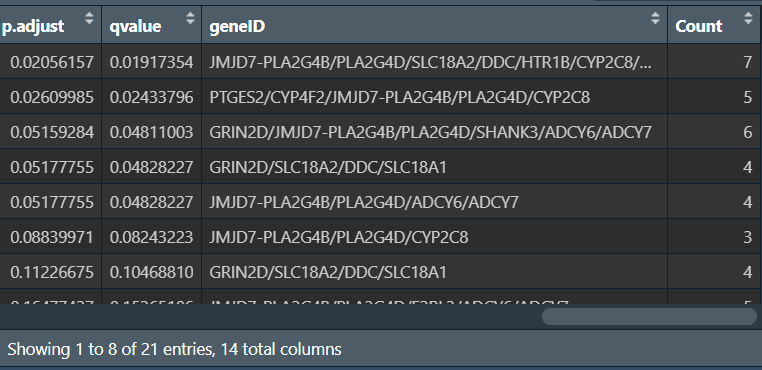
画图:
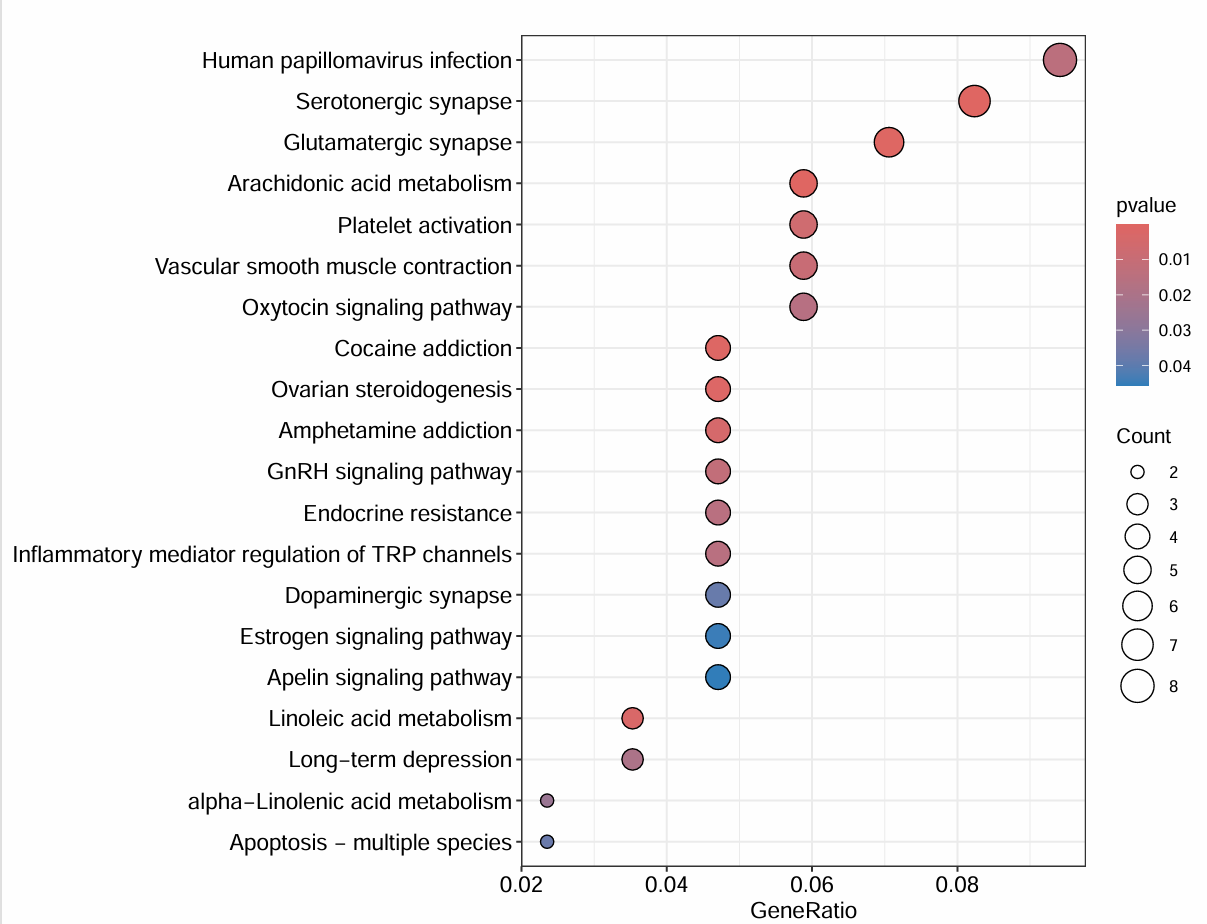
showNum <- 20; # 显示通路的数目
# 柱状图
pdf(file = "save_data\\KEGGbarplot.pdf", width = 9, height = 7);
barplot(
kk,
drop = T,
showCategory = showNum,
label_format = 130,
color = colorSel
);
dev.off();
# 气泡图
pdf(file = "save_data\\KEGGbubble.pdf", width = 9, height = 7);
dotplot(
kk,
showCategory = showNum,
orderBy = 'GeneRatio',
label_format = 130,
color = colorSel
);
dev.off();
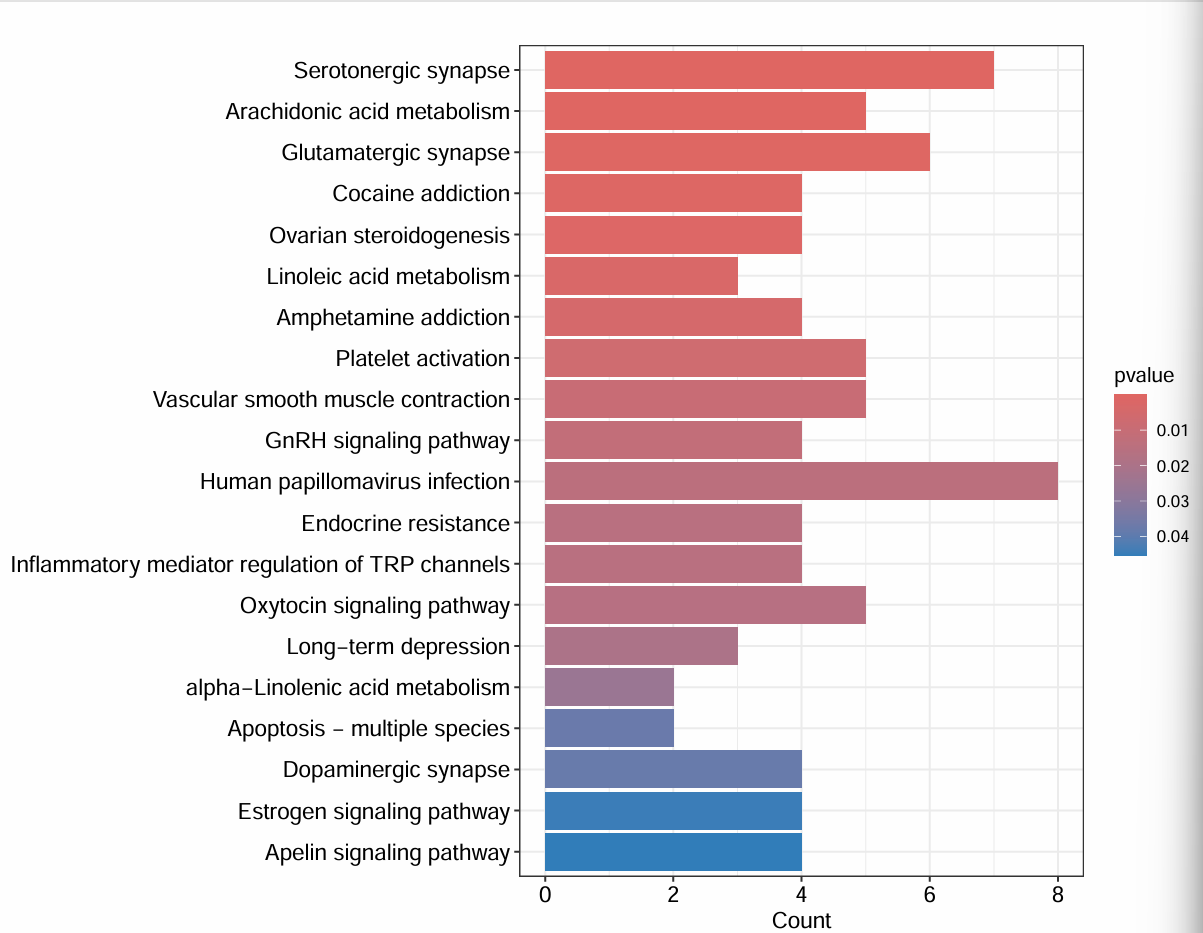
Kaplan–Meier生存分析
cox回归是分析某个基因的表达量与生存状态时间的关系,而KM生存分析是基于不同的分组来分析生存状态时间
分组方法:根据某个基因表达量的中位值,分成两组——高表达组和低表达组
需要包:survival、survminer、gridtext
if(!require("gridtext", quietly = T))
{
install.packages("gridtext");
library("gridtext");
}
library("survival");
library("survminer");
library("tidyverse");
library("readxl");
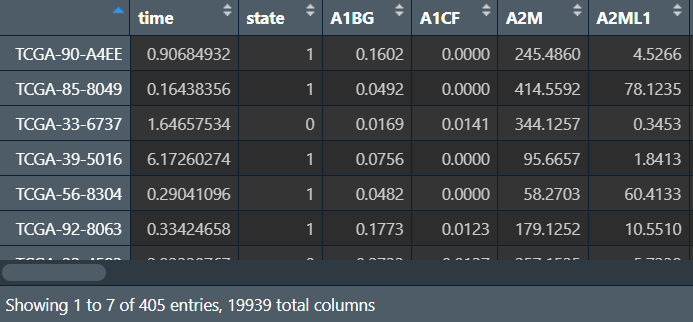
读取数据:表达矩阵和生存信息,提取共同样本后合并(同单因素cox回归)
cli <- read_excel("save_data\\time_LUSC.xlsx");
cli <- column_to_rownames(cli, "ID");
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", check.names = F, row.names = 1, sep = '\t', header = T);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
data <- t(data); # 转置
rownames(data) <- substr(rownames(data), 1, 12); # 样本名仅保留前12个字符,格式与生存信息相同
rownames(data) <- gsub('[.]', '-', rownames(data)); # 将.改为-
same_sample <- intersect(rownames(data), rownames(cli)); # 共同样本名
data <- data[same_sample, ];
cli <- cli[same_sample, ]; # 过滤
rt <- cbind(cli, data); # 合并
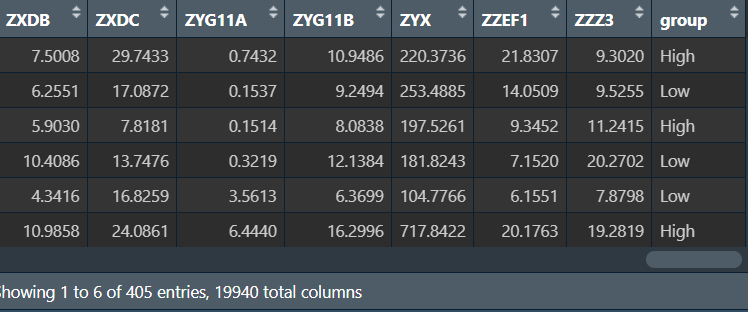
分组:这里以ABCA3基因为例
mid <- quantile(rt[, "ABCA3"], seq(0, 1, 1/2))[2]; # 中位值
rt$group <- ifelse( # 添加分组列
rt[, "ABCA3"]>mid,
"High", # 高表达组
"Low" # 低表达组
);
length = length(levels(factor(rt$group)));
生存分析:
diff <- survdiff(Surv(time, state) ~ group, data = rt);
pValue <- 1-pchisq(diff$chisq, df = length-1);
fit <- survfit(Surv(time, state) ~ group, data = rt);
生存曲线:
bioCol <- c("Firebrick3", "MediumSeaGreen", "#6E568C", "#223D6C");
bioCol <- bioCol[1:length];
surPlot <- ggsurvplot(
fit,
data = rt,
pval = paste0("p=", sprintf("%.04f", pValue)),
pval.size = 6,
legend.title = "ABCA3 expression",
legend.labs = levels(factor(rt[, "group"])),
legend = c(0.8, 0.8),
font.legend = 10,
xlab = "Time(years)",
break.time.by = 2,
palette = bioCol,
surv.median.line = "hv",
risk.table = T,
cumevents = F,
risk.table.height = 0.25
);
# 保存
pdf(file = "save_data\\KMsurvival.pdf", width = 6.5, height = 6.25, onefile = F);
print(surPlot);
dev.off();
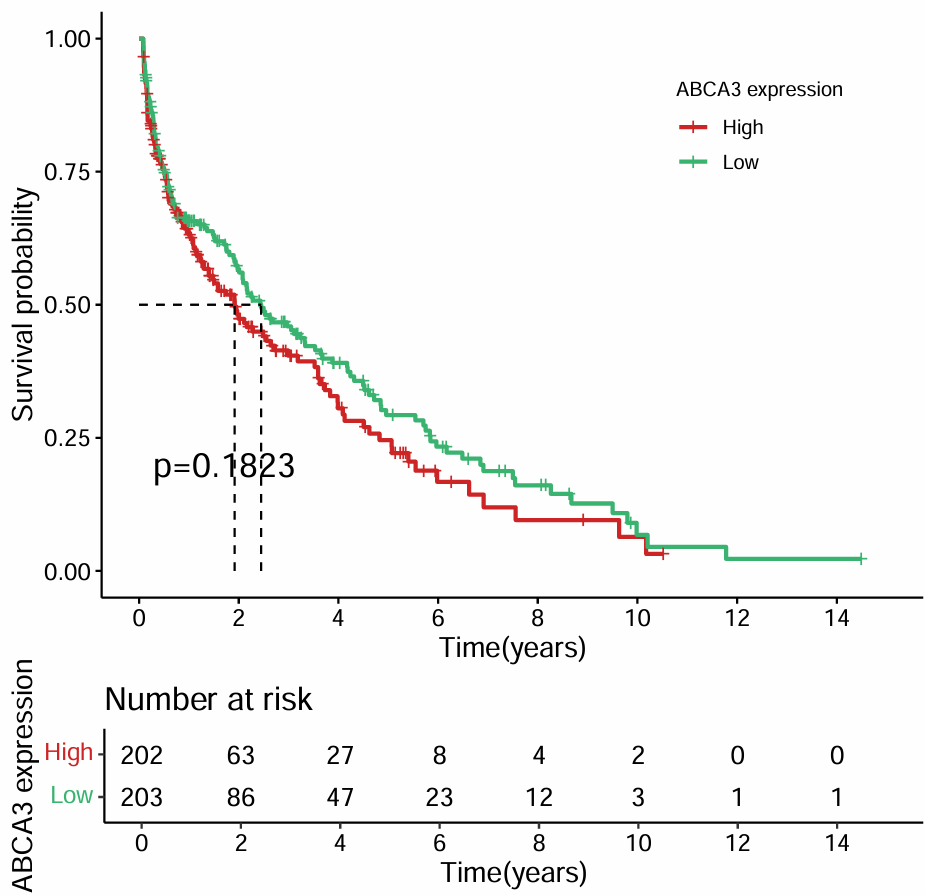
这张图展示了随着时间推移,两组患者的生存数量,图下面的数据展示了每个时间点上存活的患者数量
基因集富集分析GSEA
基因集富集分析(Gene Set Enrichment Analysis/GSEA):判断此基因集内基因的协同变化对表型变化产生的影响。它根据基因列表及logFC,不对基因进行筛选,仅按照logFC对基因排序,并以此区分纳入基因的权重
与GO/KEGG的区别:这两个都仅仅需要差异基因列表,这个列表是以某个值为阈值筛选后的,最后纳入基因的权重相同。而被舍弃的基因也可能有一定的生物学功能,GSEA就是为了这个问题
需要获取GO/KEGG的基因集:进入GSEA网站,下载以下两个文件
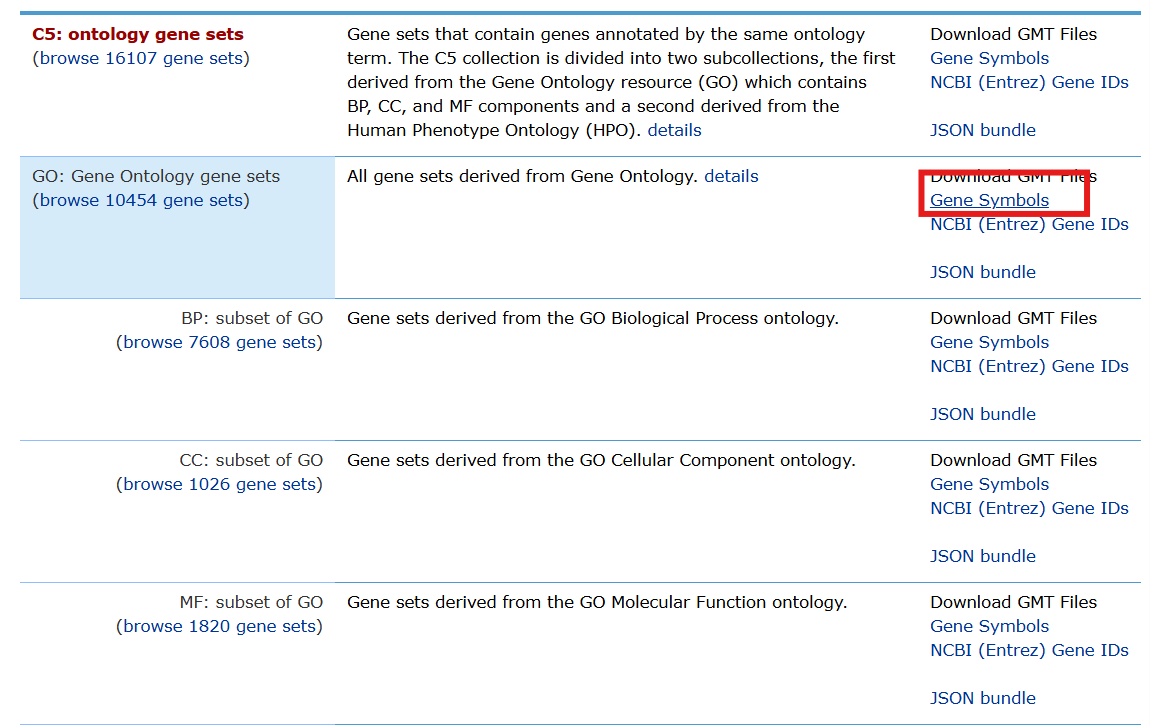

library("limma");
library("org.Hs.eg.db");
library("DOSE");
library("clusterProfiler");
library("enrichplot");
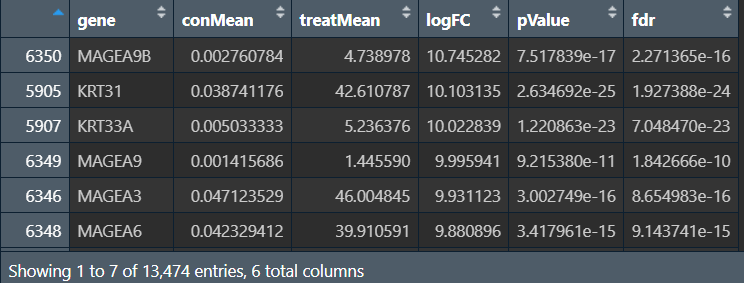
读取表达矩阵并按logFC排序,将logFC独立成一个数组,元素名为基因名:
rt <- read.table("save_data\\TCGA.all.Wilcoxon.txt", check.names = F, sep = '\t', header = T);
rt <- rt[rt[, "logFC"]!=Inf, ]; # 去除异常值
rt <- rt[order(rt[, "logFC"], decreasing = T), ]; # 排序
logFC <- as.vector(rt[, "logFC"]);
names(logFC) <- as.vector(rt[, 1]);
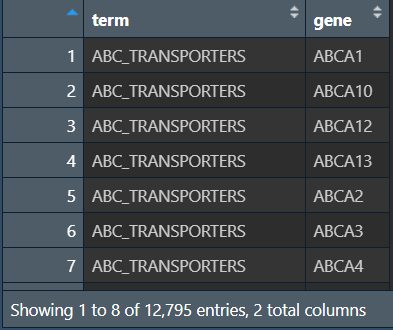
读取基因集文件:
# 使用KEGG基因集
gmt <- read.gmt("data\\GSEA\\c2.cp.kegg_legacy.v2024.1.Hs.symbols.gmt");
gmt[, 1] <- gsub("KEGG_", "", gmt[, 1]); # 去掉第一列前面的KEGG_
# 也可使用GO基因集
# gmt <- read.gmt("data\\GSEA\\c5.go.v2024.1.Hs.symbols.gmt");
# gmt[, 1] <- gsub("GO", "", gmt[, 1]); # 去掉第一列前面的GO
GSEA富集分析:
kk <- GSEA(logFC, TERM2GENE = gmt, pvalueCutoff = 1); # GSEA富集分析
kkTab <- as.data.frame(kk);
kkTab <- kkTab[kkTab$p.adjust<0.05, ]; # 根据p值筛选
write.table(kkTab, file = "save_data\\GSEA.result.KEGG.txt", row.names = F, sep = '\t', quote = F);
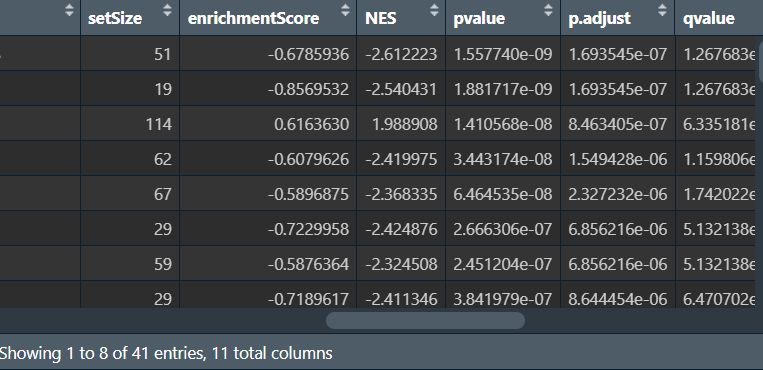
重点关注NES(富集分数)这一列,如果它<0,则该基因在正常组中显著富集;>0则在肿瘤组中显著富集
绘制肿瘤组和正常组富集图:
termNum <- 5; # 展示通路的数目
# 肿瘤组
kkUp <- kkTab[kkTab$NES>0, ];
if(nrow(kkUp)>=termNum){
showTerm <- row.names(kkUp)[1:termNum]; # 要展示的基因名称
gseaplot <- gseaplot2(
kk, # 富集分析结果
showTerm, # 要展示的基因名称
base_size = 8,
title = "Enriched in Tumor",
pvalue_table = T
);
pdf(file = "save_data\\GSEA.tumor.KEGG.pdf", width = 13, height = 11);
print(gseaplot);
dev.off();
}
# 正常组
kkDown <- kkTab[kkTab$NES<0, ];
if(nrow(kkDown)>=termNum){
showTerm <- row.names(kkDown)[1:termNum]; # 要展示的基因名称
gseaplot <- gseaplot2(
kk, # 富集分析结果
showTerm, # 要展示的基因名称
base_size = 8,
title = "Enriched in Normal",
pvalue_table = T
);
pdf(file = "save_data\\GSEA.normal.KEGG.pdf", width = 16, height = 11);
print(gseaplot);
dev.off();
}
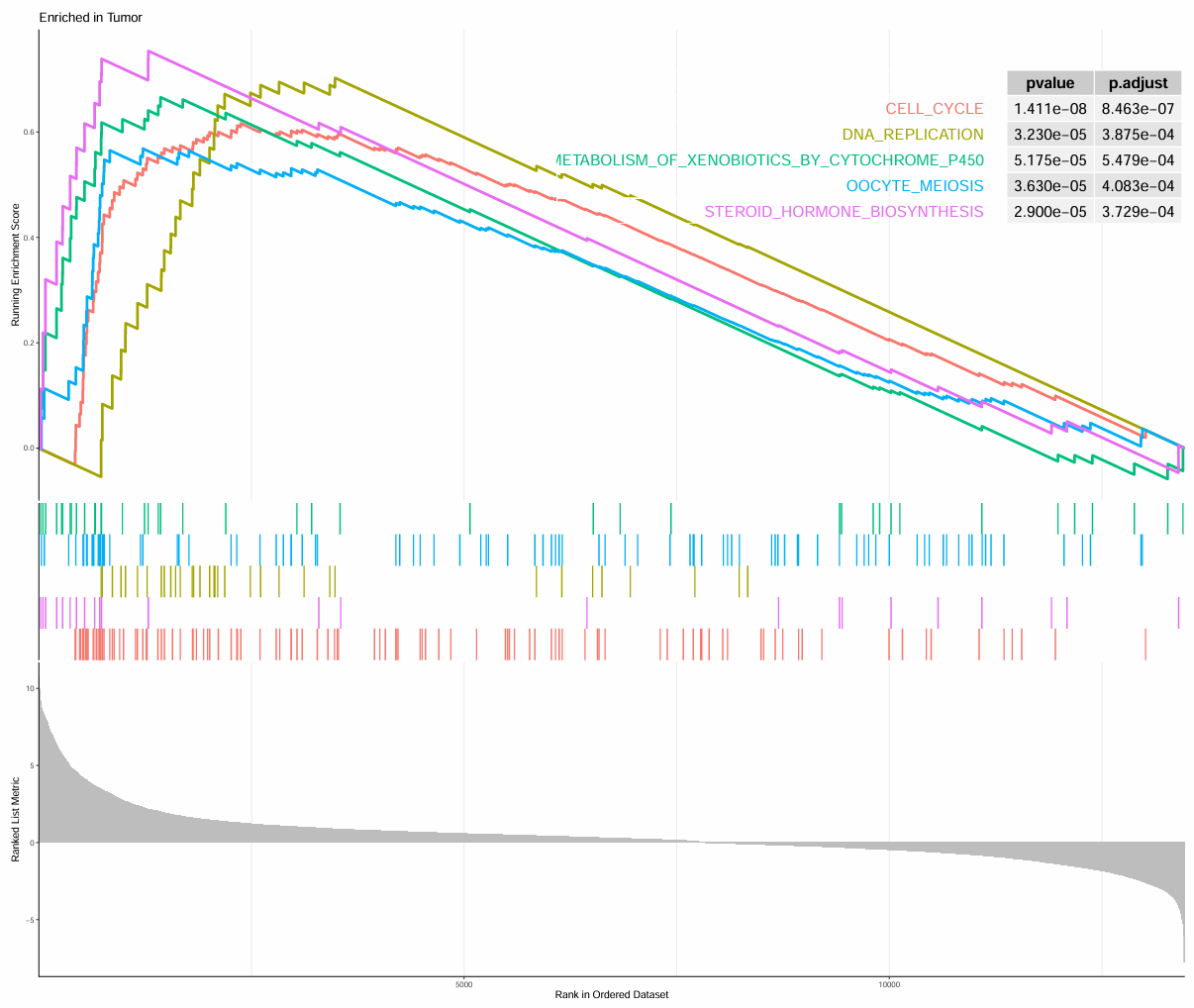
肿瘤组:
正常组:
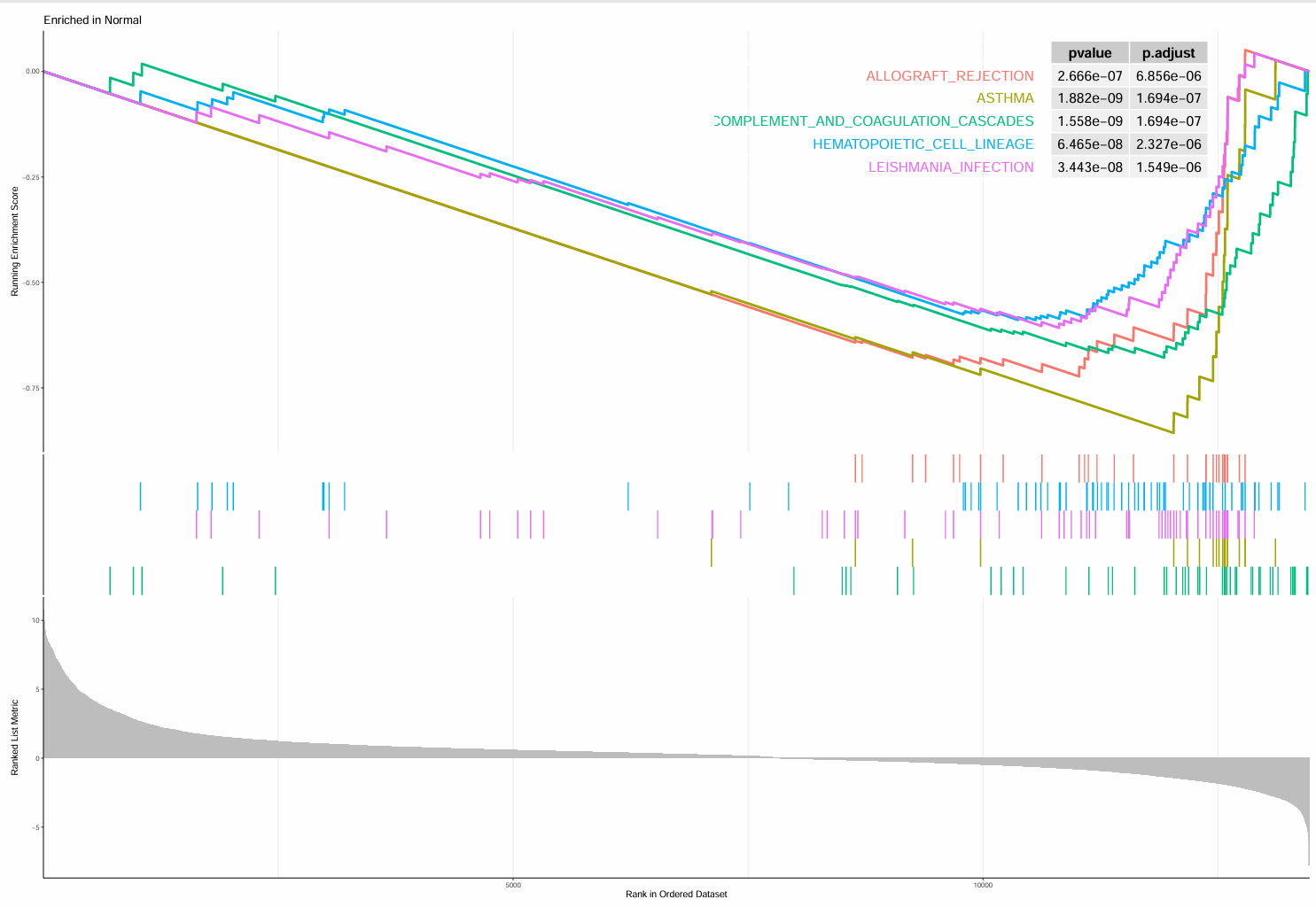
图共分为上中下三部分:
-
最上面的Enrichment score折线图:展示基因集中基因按logFC排序计算时,富集分数在计算到每个位置时的展示。折线最高/低峰的得分就是基因集的富集评分,位于最高峰前/最低峰后的的基因就是核心基因(对富集得分贡献最大的基因成员)
-
中间的hits图/条形码图:该通路基因集中成员出现在基因排序列表中的位置
-
排序后所有基因rank值的分布,L组基因高表达,H组基因低表达,灰色面积图展示每个基因的信噪比(排序值计算方式)
ssGSEA和GSVA
ssGSEA:可以理解成是单样本的GSEA,对于一个基因集S,每一个样本都可以计算得到一个NES。根据单个样本的基因表达量进行排序
GSVA与ssGSEA相同,只是使用了核密度估计的方法,用分布函数值代替基因表达值
总的来说,ssGSEA基于秩和统计评估基因集的富集程度;GSVA评估基因集的活跃程度,是非参数化的
需要包:GSEABase、GSVA
library("limma");
if(!require("GSEABase", quietly = T))
{
library("BiocManager");
BiocManager::install("GSEABase");
library("GSEABase");
}
if(!require("GSVA", quietly = T))
{
library("BiocManager");
BiocManager::install("GSVA");
library("GSVA");
}
library(pheatmap);
library(reshape2);
library(ggpubr);
读取tpm表达矩阵和基因集(这里还是以KEGG基因集为例)
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", check.names = F, sep = '\t', header = T, row.names = 1);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames); # 转为矩阵
geneSets <- getGmt(
"data\\GSEA\\c2.cp.kegg_legacy.v2024.1.Hs.symbols.gmt",
geneIdType = SymbolIdentifier()
);
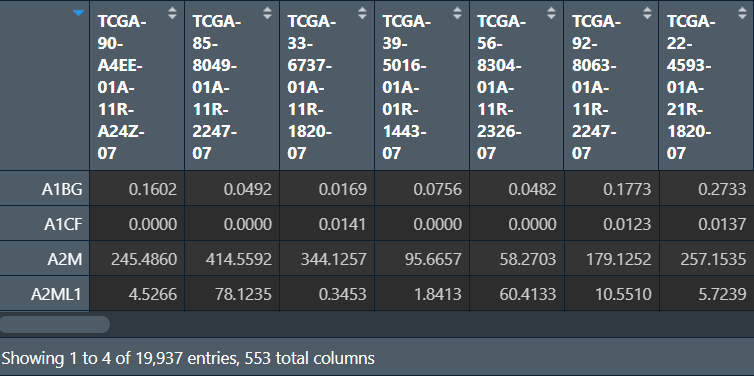
ssgsea:
# ssgsea分析
ssgsea_data <- ssgseaParam(data, geneSets);
ssgsea_res <- gsva(ssgsea_data);
# 对打分标准化
normalize <- function(x){
return((x-min(x))/(max(x)-min(x)));
}
ssgsea_res <- normalize(ssgsea_res);
# 保存结果
ssgsea_save <- rbind(id = colnames(ssgsea_res), ssgsea_res);
write.table(ssgsea_save, file = "save_data\\ssgseaOut.txt", row.names = F, sep = '\t', quote = F);
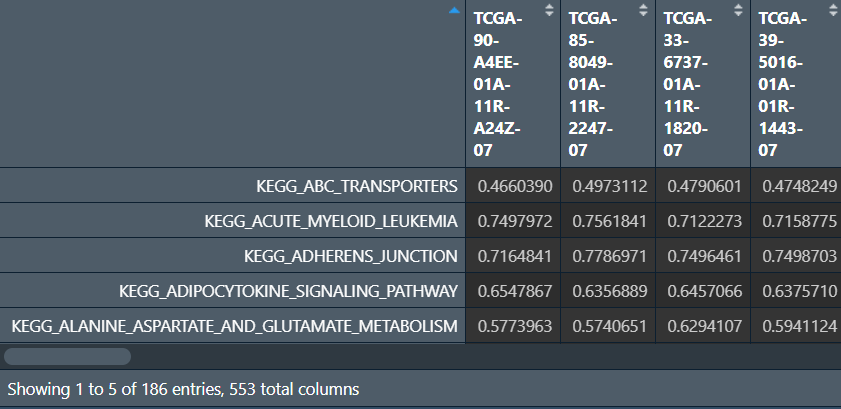
画图:先分成肿瘤组和正常组,方法同前(按样本名第14-15个字符)
# group:标识每个样本是正常还是癌症
group <- sapply(strsplit(colnames(data), '\\-'), "[", 4);
group <- sapply(strsplit(group, ''), "[", 1);
group <- gsub("2", "1", group);
con_num <- length(group[group==1]); # 正常组数量
treat_num <- length(group[group==0]); # 肿瘤组数量
# 获取新分组信息type,正常组为1, 肿瘤组为2
type <- c(rep(1, con_num), rep(2, treat_num));
# 对ssgsea_res重排
ssgsea_res1 <- ssgsea_res[, group==1];
ssgsea_res2 <- ssgsea_res[, group==0];
ssgsea_res_new <- cbind(ssgsea_res1, ssgsea_res2);
ssgsea_res_new <- cbind(type, t(ssgsea_res)); # 添加分组信息
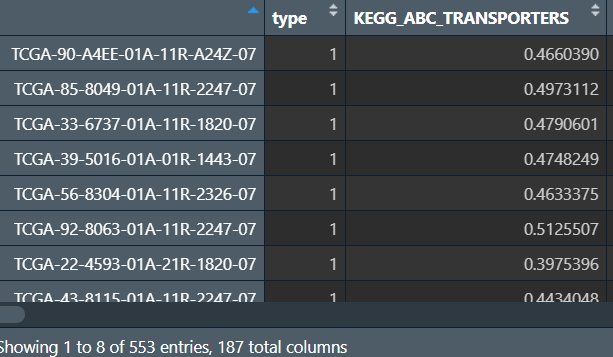
此时行名是样本名,列名是通路名
提取差异显著的通路:
sig_gene <- c(); # 差异显著的基因
for (i in colnames(ssgsea_res_new)[2:(ncol(ssgsea_res_new))]) {
test <- wilcox.test(ssgsea_res_new[, i] ~ ssgsea_res_new[, "type"]);
p <- test$p.value; # 获取p值
if(p<0.05){
sig_gene <- c(sig_gene, i); # 添加结果
}
}
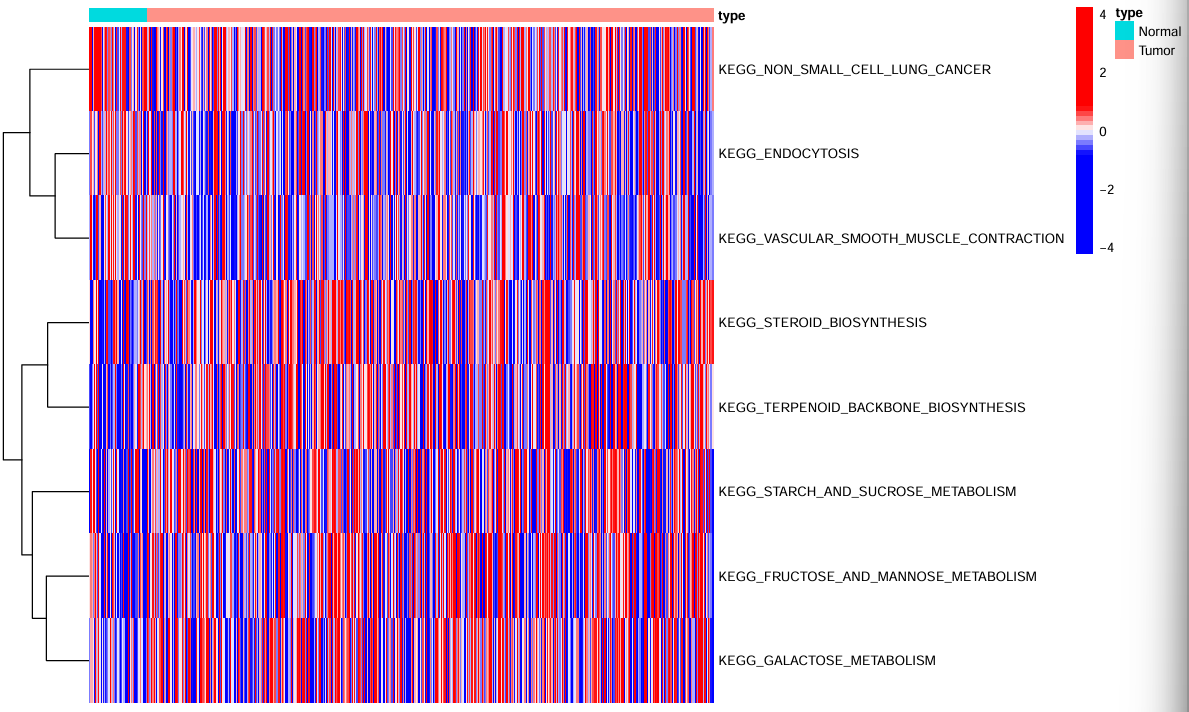
绘制热图:
hm_exp <- ssgsea_res_new[, sig_gene]; # 差异显著的通路
hm_exp <- t(hm_exp);
type <- c(rep("Normal", con_num), rep("Tumor", treat_num));
names(type) <- rownames(ssgsea_res_new);
type <- as.data.frame(type); # 样本组别
# 绘图
pdf(file = "save_data\\ssgsea_heatmap.pdf", width = 10, height = 6);
pheatmap(
hm_exp,
annotation = type,
color = colorRampPalette(c(rep("blue", 5), "white", rep("red", 5)))(50),
cluster_cols = F,
show_colnames = F,
scale = "row",
fontsize = 8,
fontsize_row = 8,
fontsize_col = 15
);
dev.off();
样本配对的差异表达分析
即寻找既测了正常又测了肿瘤的样本,对这些样本进行差异表达分析
library(limma);
library(ggpubr);
读取tpm表达矩阵,并提取正常和肿瘤组样本的表达量:
# tpm表达矩阵
tpm <- read.table("save_data\\TCGA_LUSC_TPM.txt", check.names = F, row.names = 1, sep = '\t', header = T);
dimnames <- list(rownames(tpm), colnames(tpm));
tpm <- matrix(as.numeric(as.matrix(tpm)), nrow = nrow(tpm), dimnames = dimnames);
data <- tpm[rowMeans(tpm)>1, ]; # 去除低表达的基因
# 提取表达量
group <- sapply(strsplit(colnames(data), '\\-'), "[", 4);
group <- sapply(strsplit(group, ''), "[", 1);
normal <- data[, group!=0]; # 正常组
tumor <- data[, group==0]; # 肿瘤组
对两组数据进行处理:
-
将矩阵转置,使行名为样本名,列名为基因名
-
指定要提取的基因,只保留该基因的列
-
更改样本名格式:
TCGA-90-A4EE-01A-11R-A24Z-07->TCGA.90.A4EE(只保留前3部分名称)
最后取两组数据样本名的交集,合并两组数据:
gene <- "A2M"; # 要提取的基因名称
# 正常组
normal <- t(normal);
normal <- as.matrix(normal[, gene]);
colnames(normal) <- gene;
rownames(normal) <- gsub(
"(.*?)\\-(.*?)\\-(.*?)\\-(.*?)\\-.*",
"\\1\\-\\2\\-\\3",
rownames(normal)
);
# 肿瘤组
tumor <- t(tumor);
tumor <- as.matrix(tumor[, gene]);
colnames(tumor) <- gene;
rownames(tumor) <- gsub(
"(.*?)\\-(.*?)\\-(.*?)\\-(.*?)\\-.*",
"\\1\\-\\2\\-\\3",
rownames(tumor)
);
# 取交集
same_sample <- intersect(row.names(normal), row.names(tumor));
Normal <- normal[same_sample, ];
Tumor <- tumor[same_sample, ];
same_data <- cbind(Normal, Tumor);
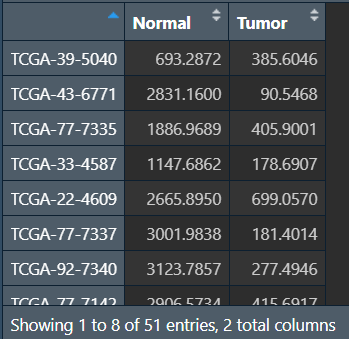
绘制图形:
pdf(file = "save_data\\single_pair_diff.pdf", width = 5.5, height = 5);
ggpaired(
as.data.frame(same_data),
cond1 = "Normal",
cond2 = "Tumor",
fill = c("red", "blue"),
palette = "jco",
xlab = "",
ylab = paste0(gene, "expression")
) +
stat_compare_means(
paired = T,
label = "p.format",
label.x = 1.35
);
dev.off();
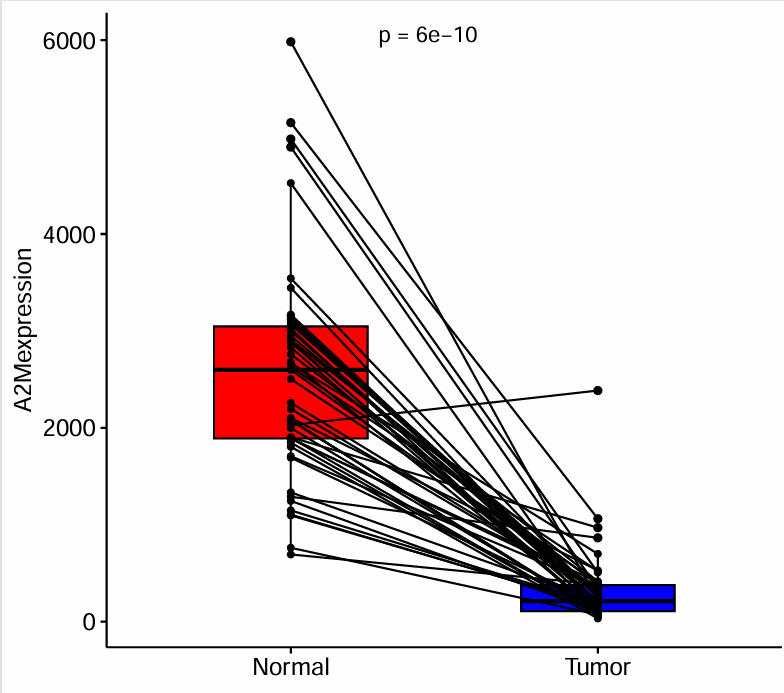
可以看到该基因在正常组中的表达量显著高于肿瘤组
对所有基因进行分析:
tpm <- read.table("save_data\\TCGA_LUSC_TPM.txt", check.names = F, row.names = 1, sep = '\t', header = T);
dimnames <- list(rownames(tpm), colnames(tpm));
tpm <- matrix(as.numeric(as.matrix(tpm)), nrow = nrow(tpm), dimnames = dimnames);
data <- tpm[rowMeans(tpm)>1, ];
group <- sapply(strsplit(colnames(data), '\\-'), "[", 4);
group <- sapply(strsplit(group, ''), "[", 1);
normal <- data[, group!=0]; # 正常组
tumor <- data[, group==0]; # 肿瘤组
normal <- as.matrix(t(normal));
rownames(normal) <- gsub("(.*?)\\-(.*?)\\-(.*?)\\-(.*?)\\-.*", "\\1\\-\\2\\-\\3", rownames(normal));
tumor <- as.matrix(t(tumor));
rownames(tumor) <- gsub("(.*?)\\-(.*?)\\-(.*?)\\-(.*?)\\-.*", "\\1\\-\\2\\-\\3", rownames(tumor));
same_sample <- intersect(row.names(normal), row.names(tumor));
normal <- normal[same_sample, ];
tumor <- tumor[same_sample, ];
# 在行名(样本名)后加上normal/tumor进行标识
rownames(normal) <- paste(rownames(normal), "normal", sep = "_");
rownames(tumor) <- paste(rownames(tumor), "tumor", sep = "_");
same_data <- rbind(normal, tumor);
same_data <- as.matrix(same_data);
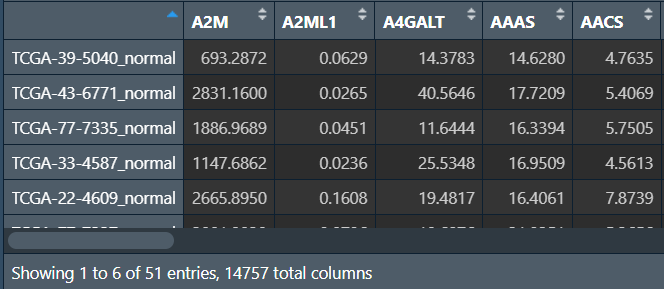
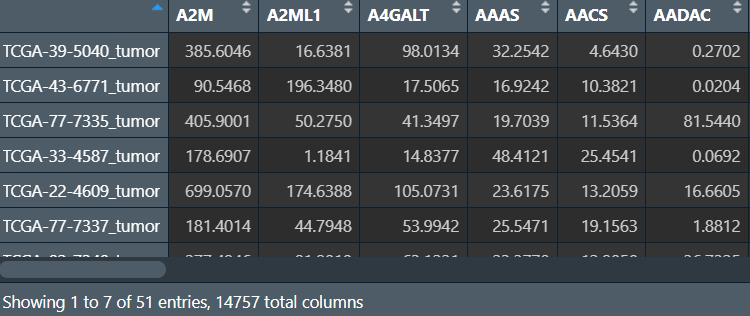
分组进行差异分析,并筛选出差异基因:(代码同差异分析那一节)
type <- c(rep(1, length(same_sample)), rep(2, length(same_sample)));
same_data <- t(same_data);
con_num <- length(same_sample);
treat_mean <- length(same_sample);
# 对每行(每个基因)进行分析
outTab <- data.frame(); # 结果矩阵
for (i in row.names(same_data)) {
rt <- data.frame(expression=same_data[i, ], type=type);
wilcoxTest <- wilcox.test(expression~type, data = rt);
pvalue <- wilcoxTest$p.value;
con_mean <- mean(same_data[i, 1:con_num]);
treat_mean <- mean(same_data[i, (con_num+1):ncol(same_data)]);
logFC <- log2(treat_mean)-log2(con_mean);
con_med <- median(same_data[i, 1:con_num]);
treat_med <- median(same_data[i, (con_num+1):ncol(same_data)]);
diffMed <- treat_med-con_med;
if((logFC>0&&diffMed>0) || (logFC<0&&diffMed<0)){
outTab <- rbind(outTab,
cbind(gene = i, conMean = con_mean, treatMean = treat_mean, logFC = logFC, pValue = pvalue));
}
}
# 计算fdr值
pvalue <- outTab[, "pValue"];
fdr <- p.adjust(as.numeric(as.vector(pvalue)), method = "fdr");
outTab <- cbind(outTab, fdr=fdr);
# 筛选差异基因
logFC_filter <- 1;
fdr_filter <- 0.05;
outDiff <- outTab[abs(as.numeric(as.vector(outTab$logFC)))>logFC_filter , ];
outDiff <- outDiff[as.numeric(as.vector(outDiff$fdr))<fdr_filter, ];
# 保存数据--差异基因
write.table(outDiff, file = "save_data\\TCGA.diff.Wilcoxon.paired.txt", sep = '\t', row.names = F, quote = F);
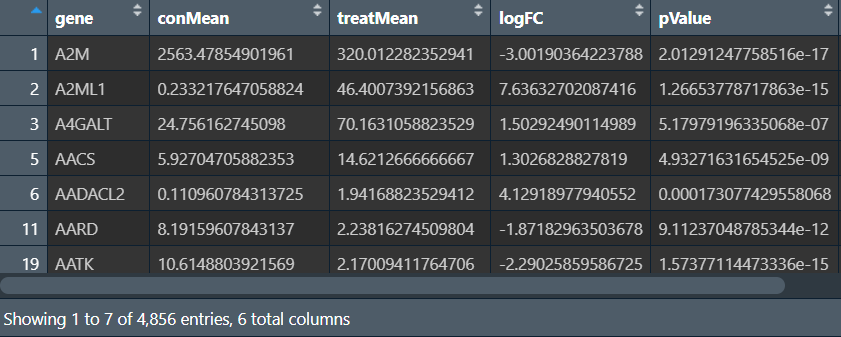
注:实际上,样本配对的差异表达分析与普通的差异表达分析 之间的差异不大
LASSO回归
在普通最小二乘法的基础上,加入一个惩罚项,通过调整惩罚项系数来降低回归系数的方差,从而减少多重共线性的影响,并防止模型过拟合
需要数据:tpm表达矩阵、生存时间和状态、单因素cox回归结果
需要包:glmnet、survival
if(!require("glmnet", quietly = T))
{
install.packages("glmnet");
install.packages("iterators");
library("glmnet");
}
library(survival);
读取数据,并提取表达矩阵和cox结果的共同基因,根据共同样本合并生存信息和表达矩阵:
library("readxl");
library("tidyverse");
# 生存信息
cli <- read_excel("save_data\\time_LUSC.xlsx");
cli <- column_to_rownames(cli, "ID");
# tpm表达矩阵
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", check.names = F, row.names = 1, sep = '\t', header = T);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
# 单因素cox
unicox_gene <- read.table("save_data\\uniCox.txt", check.names = F, row.names = 1, sep = '\t', header = T);
# 提取单因素cox基因的表达矩阵
data <- data[rownames(unicox_gene), ];
data <- t(data); # 转置
rownames(data) <- substr(rownames(data), 1, 12); # 样本名仅保留前12个字符,格式与生存信息相同
rownames(data) <- gsub('[.]', '-', rownames(data)); # 将.改为-
same_sample <- intersect(rownames(data), rownames(cli)); # 共同样本名
data <- data[same_sample, ];
cli <- cli[same_sample, ]; # 过滤
rt <- cbind(cli, data); # 合并
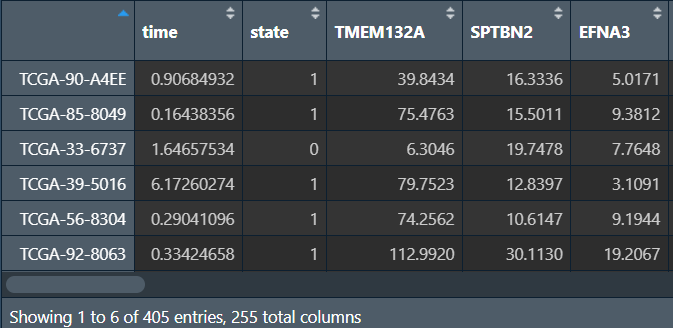
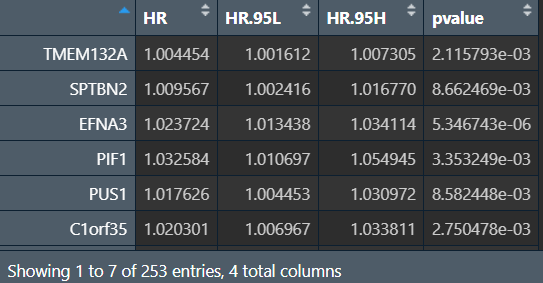
构建lasso回归模型:
-
使用
set.seed设置随机种子,使K折交叉验证结果固定 -
glmnet函数的family参数取值:、-
"gaussian"一维连续因变量 -
"mgaussian"多维连续因变量 -
"poison"非负次数因变量 -
"binomial"二元离散因变量 -
"multinomial"多元离散因变量
-
set.seed(123456); # 设置随机种子
x <- as.matrix(rt[, c(3:ncol(rt))]); # 每个样本的各基因表达量
y <- data.matrix(Surv(rt$time, rt$state)); # 生存信息
fit <- glmnet(x, y, family = "cox", nfolds = 10); # 构建模型
绘图:
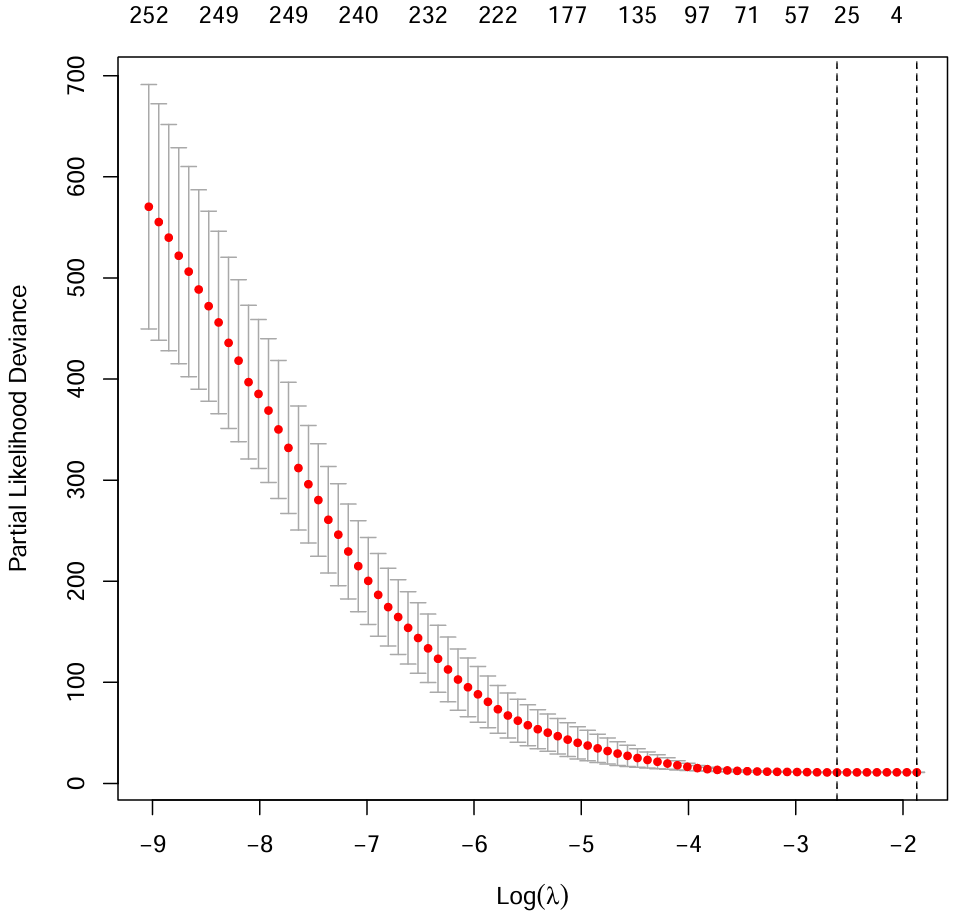
# c-index(交叉验证曲线)
cvfit <- cv.glmnet(x, y, family = "cox", type.measure = "C", nfolds = 10);
pdf(file = "save_data\\lasso.c-index.pdf");
plot(cvfit);
abline(v = log(c(cvfit$lambda.min, cvfit$lambda.1se)), lty = "dashed");
dev.off();
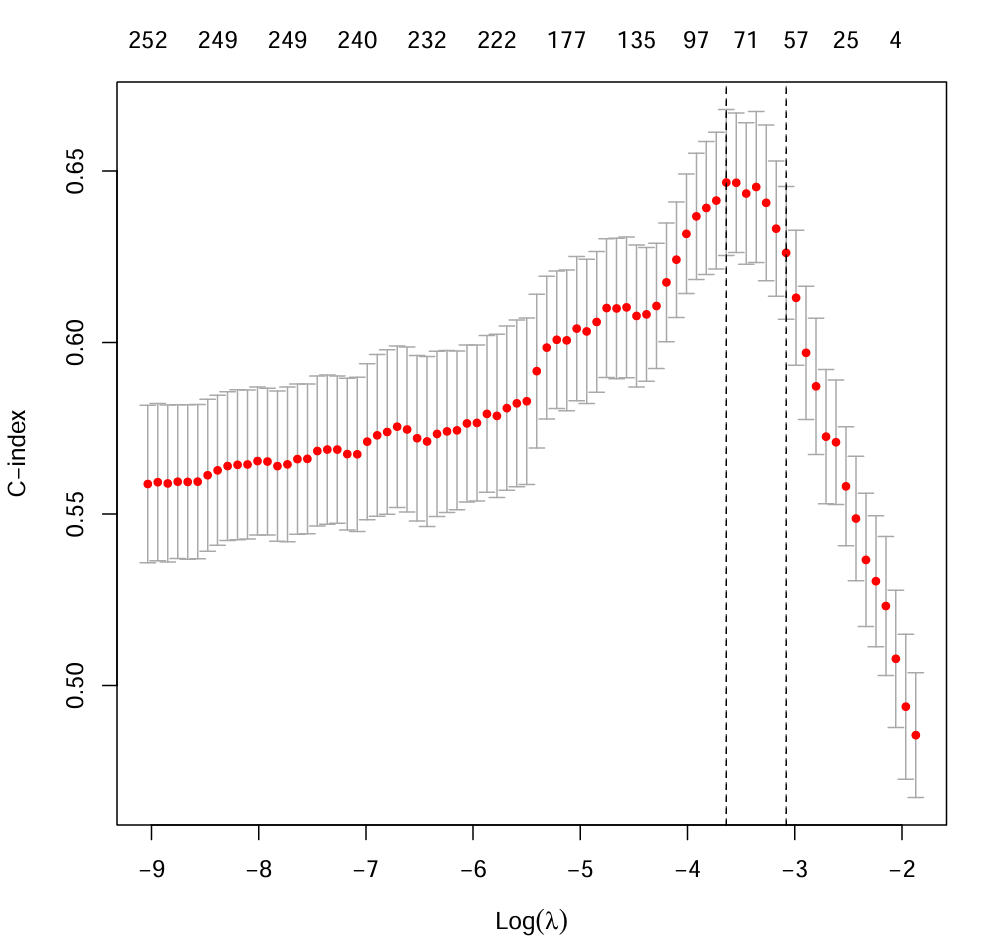
# deviance(偏似然偏差)
cvfit <- cv.glmnet(x, y, family = "cox", type.measure = "deviance", nfolds = 10);
pdf(file = "save_data\\lasso.cvfit.pdf");
plot(cvfit);
abline(v = log(c(cvfit$lambda.min, cvfit$lambda.1se)), lty = "dashed");
dev.off();
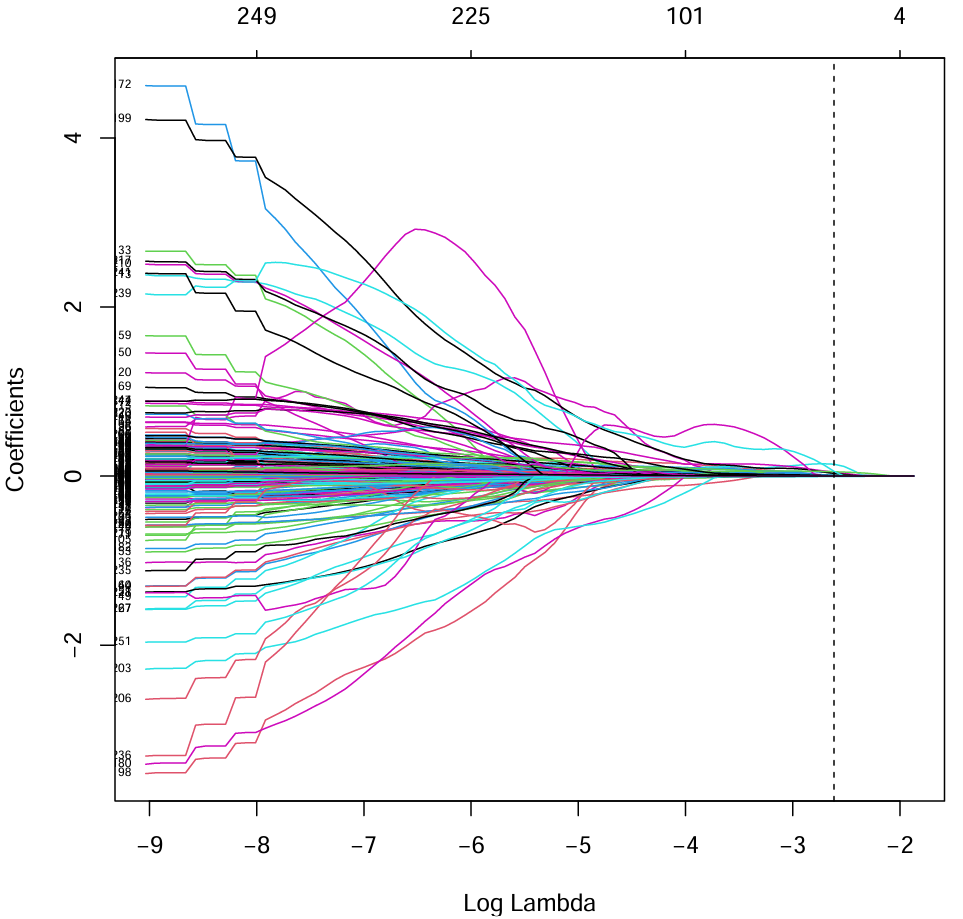
# coefficients(回归系数路径图)
pdf(file = "save_data\\lasso.lambda.pdf");
plot(fit, xvar = "lambda", label = T);
abline(v = log(cvfit$lambda.min), lty = "dashed");
dev.off();
lasso回归结果:
coef <- coef(fit, s = cvfit$lambda.min);
index <- which(coef!=0); # 筛选系数不为0的基因为lasso回归结果
act_coef <- coef[index]; # 每个基因对应的系数
lasso_gene <- row.names(coef)[index]; # lasso回归基因
lasso_res <- data.frame(
gene = lasso_gene,
coef = act_coef
);
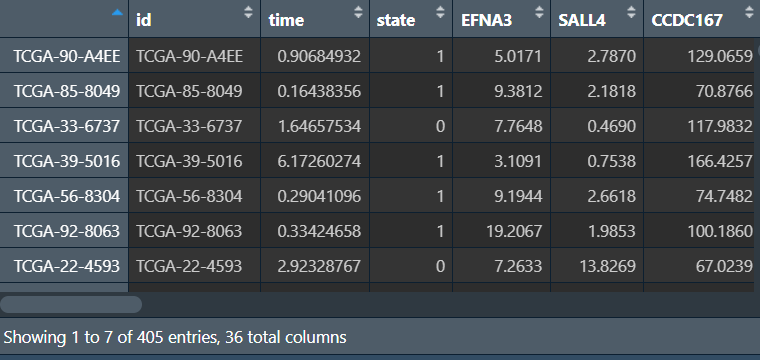
lasso_sig_exp <- rt[, c("time", "state", lasso_gene)];
lasso_sig_exp_save <- cbind(
id = row.names(lasso_sig_exp),
lasso_sig_exp
);
write.table(lasso_sig_exp_save, file = "save_data\\lasso.SigExp.txt", row.names = F, sep = '\t', quote = F);
lasso_res(lasso回归基因名及对应系数):
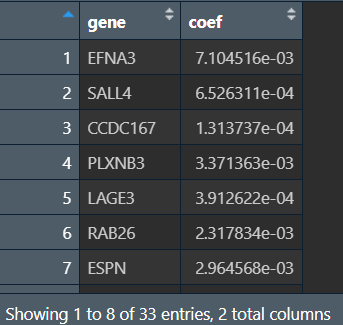
可以看到共有33个基因被筛选出来
lasso_sig_exp_save(lasso回归基因在各样本的表达量及生存信息):
多因素cox回归
以生存状态和生存时间为因变量,同时分析多因素对生存期的影响。可分析带有截尾生存时间(生存时间的截止不是由于死亡,而是其它原因引起)的资料,且不要求估计资料的生存分布类型
使用数据:上面得到的lasso回归基因在各样本的表达量及生存信息
加载包并读取数据:
library(survival);
rt <- read.table("save_data\\lasso.SigExp.txt", check.names = F, row.names = 1, sep = '\t', header = T);
构建cox模型:如果基因数>20,就使用逐步回归方式step(cox模型, direction = "both")(可选参数:”both”/”backward”/”forward”),它可以进一步筛选影响较大的基因;反之就不执行该行代码
# 构建模型
multi_cox <- coxph(Surv(time, state) ~ ., data = rt);
multi_cox <- step(multi_cox, direction = "both");
multi_cox_sum <- summary(multi_cox);
# 输出结果
outTab <- data.frame();
outTab <- cbind(
coef = multi_cox_sum$coefficients[, "coef"],
HR = multi_cox_sum$conf.int[, "exp(coef)"],
HR.95L = multi_cox_sum$conf.int[, "lower .95"],
HR.95H = multi_cox_sum$conf.int[, "upper .95"],
pvalue = multi_cox_sum$coefficients[, "Pr(>|z|)"]
);
outTab <- cbind(id = row.names(outTab), outTab);
outTab <- gsub("`", "", outTab);
write.table(outTab, file = "save_data\\multiCox.txt", row.names = F, sep = '\t', quote = F);
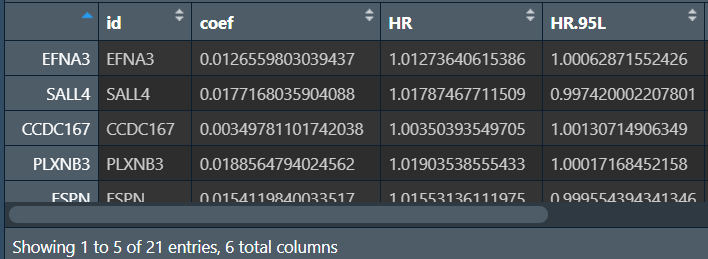
共筛选出21个基因
计算病人风险值:
risk_score <- predict(multi_cox, type = "risk", newdata = rt);
cox_gene <- rownames(multi_cox_sum$coefficients);
cox_gene <- gsub("`", "", cox_gene);
col_name <- c("time", "state", cox_gene);
risk <- as.vector(ifelse(
risk_score>median(risk_score), # 以风险得分中值为分界线
"high", "low" # 分成高低风险两类
));
# 风险计算结果:每个样本的生存信息、筛选后基因表达量、风险得分、风险高低
risk_res <- cbind(
riskScore = risk_score,
risk,
rt[, col_name]
);
risk_res_save <- cbind(id = row.names(risk_res), risk_res);
write.table(risk_res_save, file = "save_data\\risk.txt", row.names = F, sep = '\t', quote = F);
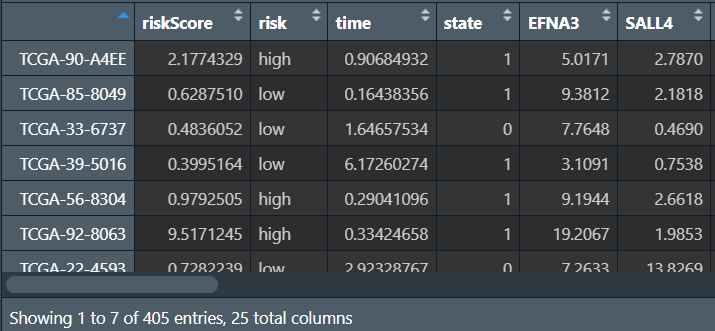
绘图(同单因素cox回归):
rt <- read.table( "save_data\\multiCox.txt", check.names = F, row.names = 1, sep = '\t', header = T);
gene <- rownames(rt);
hr <- sprintf("%.3f", rt$HR);
hrLow <- sprintf("%.3f", rt$HR.95L);
hrHigh <- sprintf("%.3f", rt$HR.95H);
Hazard.ratio <- paste0(hr, "(", hrLow, "-", hrHigh, ")");
pVal <- ifelse(rt$pvalue<0.001, "<0.001", sprintf("%.3f", rt$pvalue));
n <- nrow(rt);
nRow <- n+1;
ylim <- c(1, nRow);
layout_matrix <- matrix(c(1, 2), nc=2);
pdf(file = "save_data\\multiCoxforest.pdf", width = 7, height = nrow(rt)/13+5);
layout(layout_matrix, width = c(3, 2.5));
xlim <- c(0, 3);
par(mar=c(4, 2.5, 2, 1));
plot(1, xlim = xlim, ylim = ylim, type = "n", axes = F, xlab = "", ylab = "" );
text.cex <- 0.8;
text(0, n:1, gene, adj=0, cex=text.cex);
text(1.4, n:1, pVal, adj=1, cex=text.cex);
text(1.4, n+1, 'pvalue', adj=1, cex=text.cex, font=2);
text(3, n:1, Hazard.ratio, adj=1, cex=text.cex);
text(3, n+1, 'Hazard ratio', adj=1, cex=text.cex, font=2);
xlim <- c(0, max(as.numeric(hrLow), as.numeric(hrHigh)));
par(mar=c(4, 1, 2, 1), mgp=c(2, 0.5, 0));
plot(1, xlim = xlim, ylim = ylim, type = "n", axes = F, xlab = "Hazard ratio", ylab = "", xaxs = "i");
abline(v=1, col="black", lty=2, lwd=2);
arrows(as.numeric(hrLow), n:1, as.numeric(hrHigh), n:1, angle = 90, code = 3, length = 0.05, col = "darkblue", lwd = 2.5);
boxcolor <- ifelse(as.numeric(hr)>1, "red", "blue");
points(as.numeric(hr), n:1, pch = 15, col = boxcolor, cex = 1.6);
axis(1);
dev.off();