b站生信课程02-3
r-bioinfolessonb站生信课程TCGA及GEO数据挖掘入门必看P18-P26笔记——样本聚类、ROC曲线、线上画Venn图、列线图、免疫组化图片、免疫细胞浸润分析、免疫功能分析
写在前面:本篇教程来自b站课程TCGA及GEO数据挖掘入门必看 P18-P26 相关资料下载
一致性聚类与无监督聚类
需要数据:多因素cox回归结果(其实也可以是lasso/单因素cox回归结果)、tpm表达矩阵
需要包:ConsensusClusterPlus
if(!require("ConsensusClusterPlus", quietly = T))
{
library("BiocManager");
BiocManager::install("ConsensusClusterPlus");
library("ConsensusClusterPlus");
}
读取数据:选出肿瘤组的样本,取出筛选后基因的在各样本中的表达量
# tpm表达矩阵
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", check.names = F, row.names = 1, sep = '\t', header = T);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
# 选出肿瘤组的样本
group <- sapply(strsplit(colnames(data), '\\-'), "[", 4);
group <- sapply(strsplit(group, ''), "[", 1);
data <- data[, group==0];
# 筛选基因
multi_cox_gene <- read.table("save_data\\multiCox.txt", check.names = F, row.names = 1, sep = '\t', header = T);
data <- data[rownames(multi_cox_gene), ];
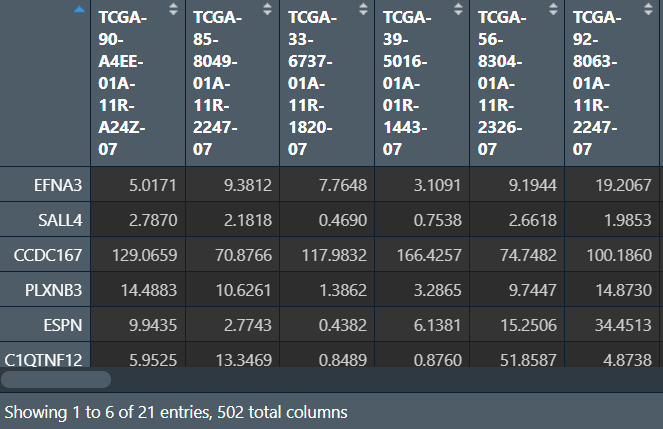
对样品进行聚类分型,使用ConsensusClusterPlus函数:
-
maxK最大的K值,形成一系列梯度 -
pItem选择百分之多少的样本重复抽样 -
pfeature选择百分之多少的基因重复抽样 -
reps重复抽样的数目,可以先设置为100,结果不错再设置为1000(这样结果更严谨) -
clusterAlg聚类算法,取值:”hc”/”pam”/”km” -
distanc距离矩阵算法,取值:”pearson”/”spearman”/”euclidean” -
title输出结果的文件夹名字,包含输出的图片等 -
seed随机种子,用于固定结果 -
plot输出图片的格式
res <- ConsensusClusterPlus(
data,
maxK = 9,
reps = 100,
pItem = 0.8,
pFeature = 1,
title = "save_data\\ConsensusClusterPlus",
clusterAlg = "pam",
distance = "euclidean",
seed = 123,
plot = "png"
);
结果分析:
-
第一张图标识相关度与颜色的关系(图例):1是非常相关(蓝色),0是不相关(白色)
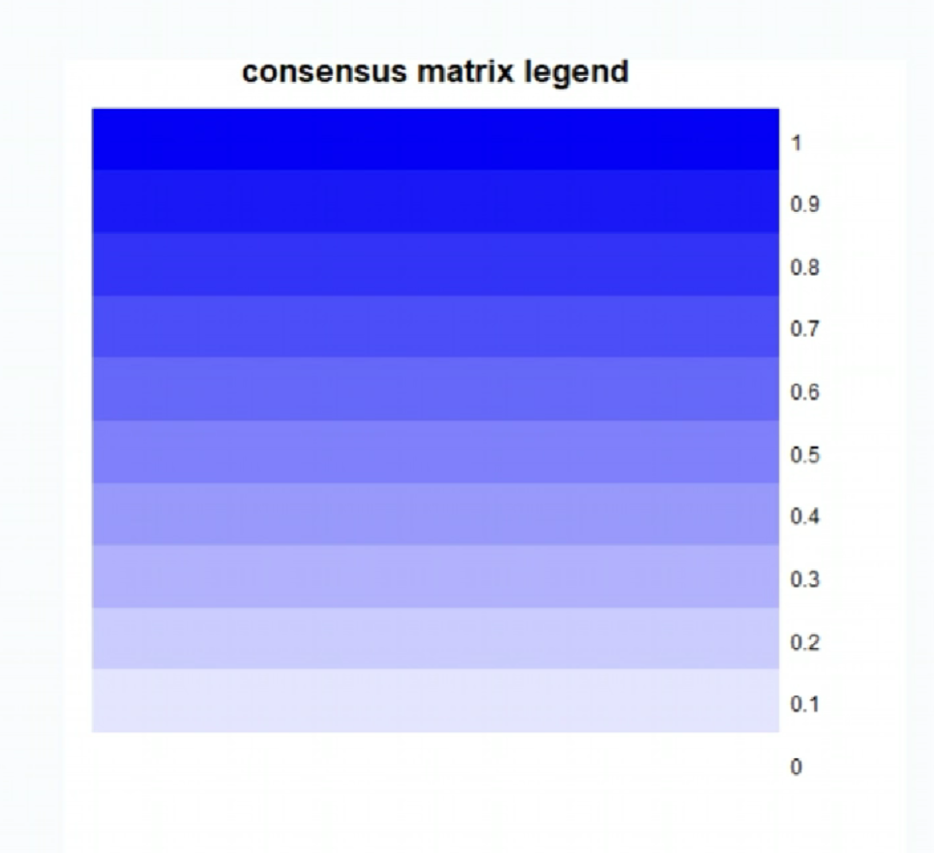
-
002-009:每个k值(分成了多少组)对应的聚类结果
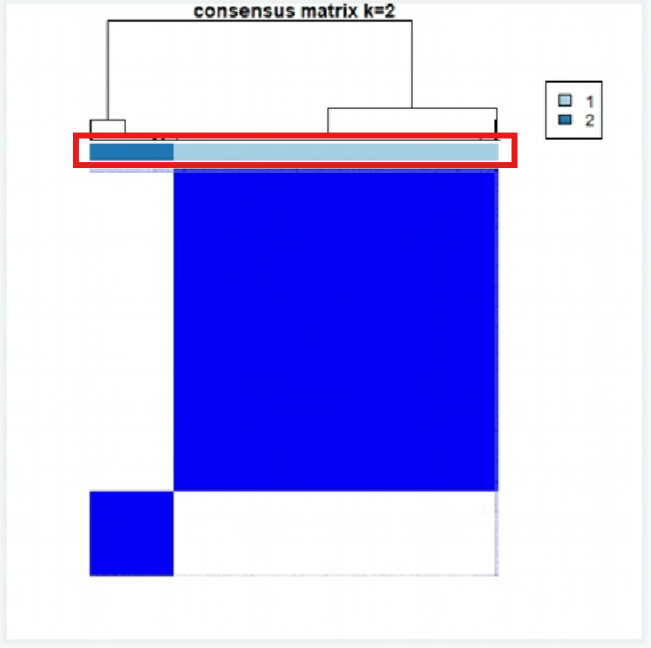
红框部分的不同颜色代表不同的组,它下面的2*2个方块代表每组的差异
评判标准(以k=2为例):
-
组内的差异小(右上和左下两个块足够蓝)
-
组间的差异大(右下和左上两个块足够白)
-
每组的样本数不能过小(不能小于总样本的10%),可以通过红框中颜色占比看出
可以看到k=2的图是符合标准的
-
-
010CDF值:
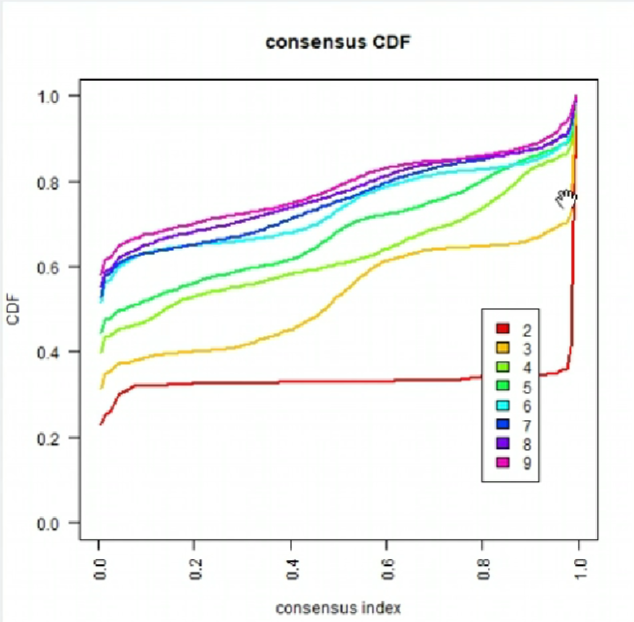
曲线在x=0.1~0.9的变化越小的越好。可以看出k=2(红色线)符合标准
-
011CDF值变化:
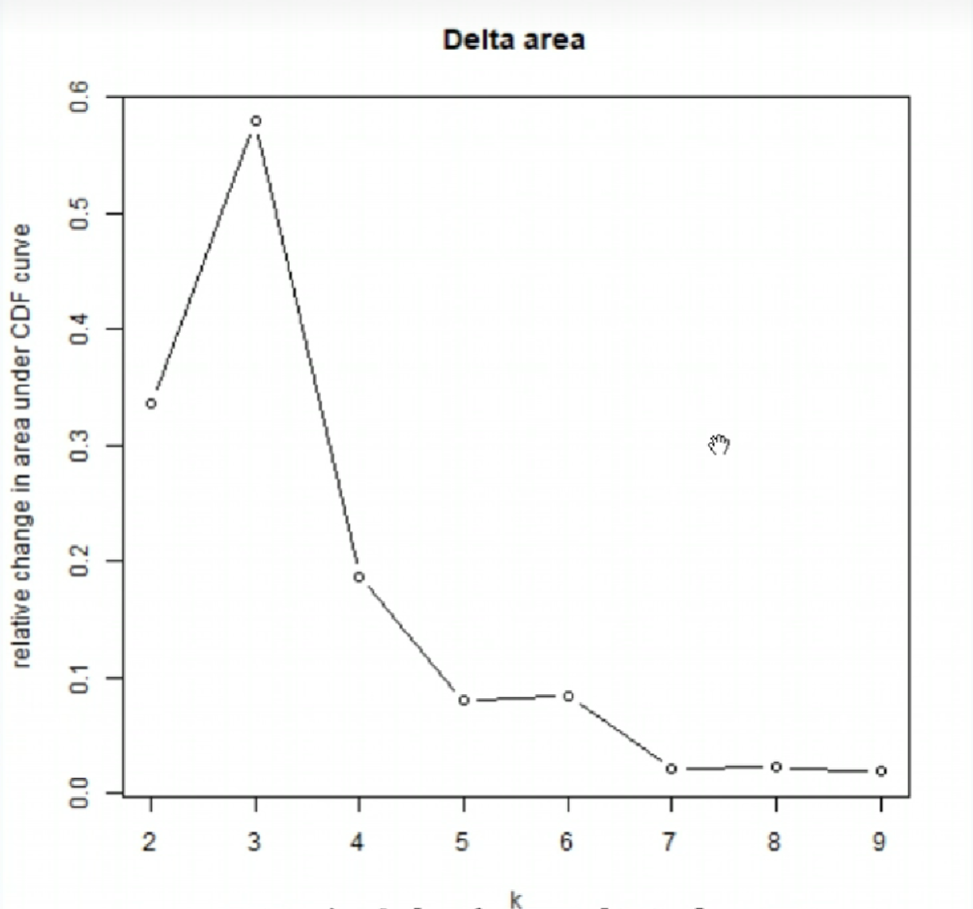
一般选取折线拐点(曲线变化趋势改变最大的点)附近的点,该图中拐点为x=3
综合上面的分析,我们选取k=2的结果
根据上面的分组,对样本进行分型:
clu_num <- 2; # 分成几组(k值)
clu <- res[[clu_num]][["consensusClass"]]; # 聚类结果(分组信息)
clu <- as.data.frame(clu);
colnames(clu) <- c("cluster");
letter <- LETTERS[1:10]; # 每组的名称,这里是ABCD大写字母
uniq_clu <- levels(factor(clu$cluster)); # 原来每组的名称
clu$cluster <- letter[match(clu$cluster, uniq_clu)]; # 将每组名称改成我们刚才定义的大写字母
clu_save <- rbind(ID = colnames(clu), clu);
write.table(clu_save, file = "save_data\\cluster.txt", sep = '\t', row.names = F, quote = F);
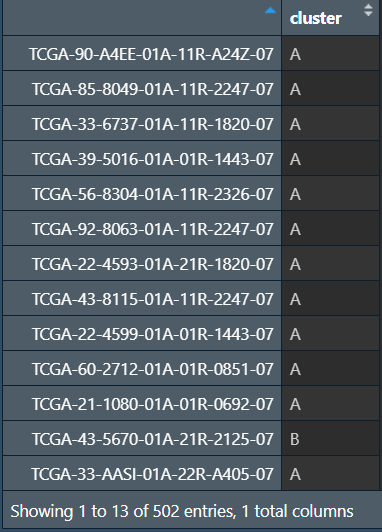
可以看到样品被分为了AB两组
ROC曲线
横坐标1-Specificity是特异性(假阳性概率),纵坐标Sentivity是敏感性(真阳性概率)
AUC指曲线下的面积
-
AUC=1:是完美的分类器
-
0.5< AUC <1:优于随机猜测,数值越大越好
-
AUC=0.5:等同于随机猜测,没有预测价值
-
AUC<0.5:比随机猜测差。但如果是反向猜测,该模型也可能优于随机猜测
if(!require("pROC", quietly = T))
{
library("BiocManager");
BiocManager::install("pROC");
library(pROC);
}
library(survival);
library(survminer);
if(!require("timeROC", quietly = T))
{
library("BiocManager");
install.packages('listenv');
install.packages('parallelly');
BiocManager::install("timeROC");
library(timeROC);
}
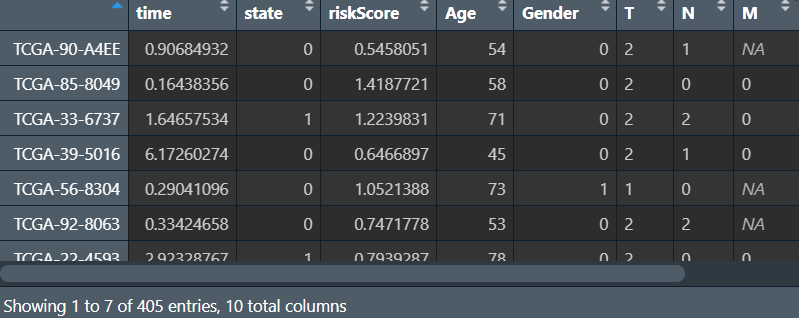
读取并处理生存信息,与风险得分合并:
# 读取生存信息
library("readxl");
library(tidyverse);
cli <- read_excel("save_data\\clinical.xlsx");
cli <- cli[, c("survival_time", "vital_status", "days_to_birth", "gender", "T", "N", "M", "stage_event", "anatomic_neoplasm_subdivision", "bcr_patient_barcode")];
# 处理生存信息
cli <- column_to_rownames(cli, "bcr_patient_barcode"); # 更改行名为样本名
cli$time <- as.numeric(cli$survival_time)/365; # 存活天数用年表示
cli$state <- ifelse(cli$vital_status=='Alive', 0, 1); # 0表示存活,1表示死亡
cli$Age <- round(as.numeric(cli$days_to_birth)/(-365)); # 年龄用年表示
cli$Gender <- ifelse(cli$gender=="MALE", 0, 1); # 性别用01表示
cli$`T` <- substr(cli$`T`, 1, 1);
cli$`N` <- substr(cli$`N`, 1, 1);
cli$`M` <- substr(cli$`M`, 1, 1); # TNM列只取第一个字符
cli$`T` <- gsub('X', NA, cli$`T`);
cli$`N` <- gsub('X', NA, cli$`N`);
cli$`M` <- gsub('X', NA, cli$`M`); # TNM列将X替换为NA
cli$stage_event <- ifelse(grepl('X', cli$stage_event), NA, cli$stage_event); # X变NA
cli$stage_event <- ifelse(grepl('IV', cli$stage_event), "4", cli$stage_event); # IV变4
cli$stage_event <- ifelse(grepl('III', cli$stage_event), "3", cli$stage_event); # III变3
cli$stage_event <- ifelse(grepl('II', cli$stage_event), "2", cli$stage_event); # II变2
cli$stage_event <- ifelse(grepl('I', cli$stage_event), "1", cli$stage_event); # I变1
cli$Stage <- as.numeric(cli$stage_event);
cli$`T` <- as.numeric(cli$`T`);
cli$`N` <- as.numeric(cli$`N`);
cli$`M` <- as.numeric(cli$`M`); # TNM、Stage列转为数值型
cli$subdivision <- ifelse( # 开头L->1 R->2
startsWith(cli$anatomic_neoplasm_subdivision, "L"),
1,
ifelse(
startsWith(cli$anatomic_neoplasm_subdivision, "R"),
2,
NA
)
);
# 读取风险得分
risk <- read.table("save_data\\risk.txt", check.names = F, row.names = 1, sep = '\t', header = T);
# 合并
same_sample <- intersect(row.names(risk), row.names(cli));
risk <- risk[same_sample, ];
cli <- cli[same_sample, ];
rt <- cbind(
cli[, c("time", "state")],
riskScore = risk[, c("riskScore")],
cli[, c("Age", "Gender", "T", "N", "M", "Stage", "subdivision")]
);
# 保存生存信息
library(writexl);
write_xlsx(cbind(ID = row.names(cli), cli[, c("time", "state", "Age", "Gender", "T", "N", "M", "Stage", "subdivision")]), "save_data\\new_clinical.xlsx");
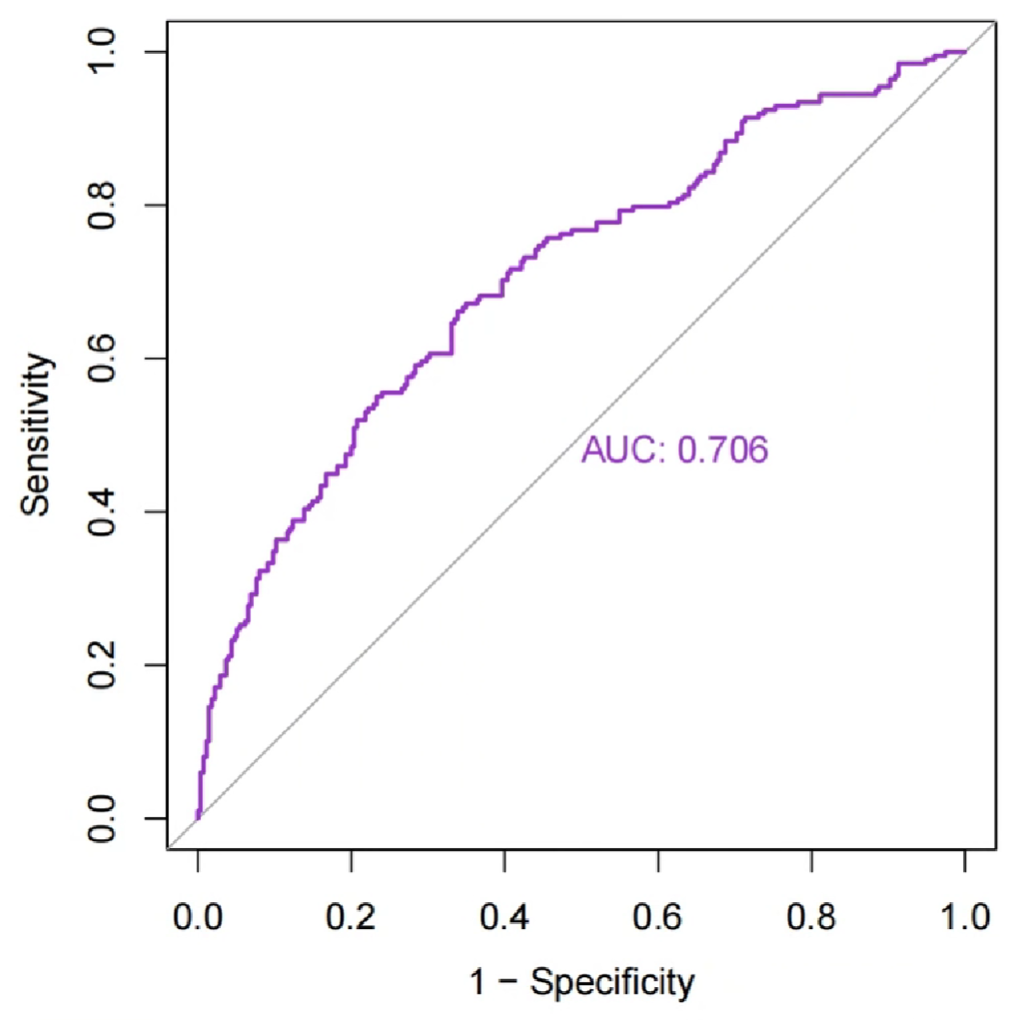
ROC分析并绘图:
# 可以是state/是否为肿瘤组~基因表达量/风险得分
roc1 <- roc(rt$state ~ rt$riskScore);
pdf(file = "save_data\\ROC.riskscore.pdf", width = 5, height = 5);
bioCol = c("DarkOrchid", "Orange2", "MediumSeaGreen", "NavyBlue", "#8B668B", "#FF4500", "#135612", "#561214");
plot(
roc1,
print.auc = T,
col = bioCol,
legacy.axes = T
);
dev.off();
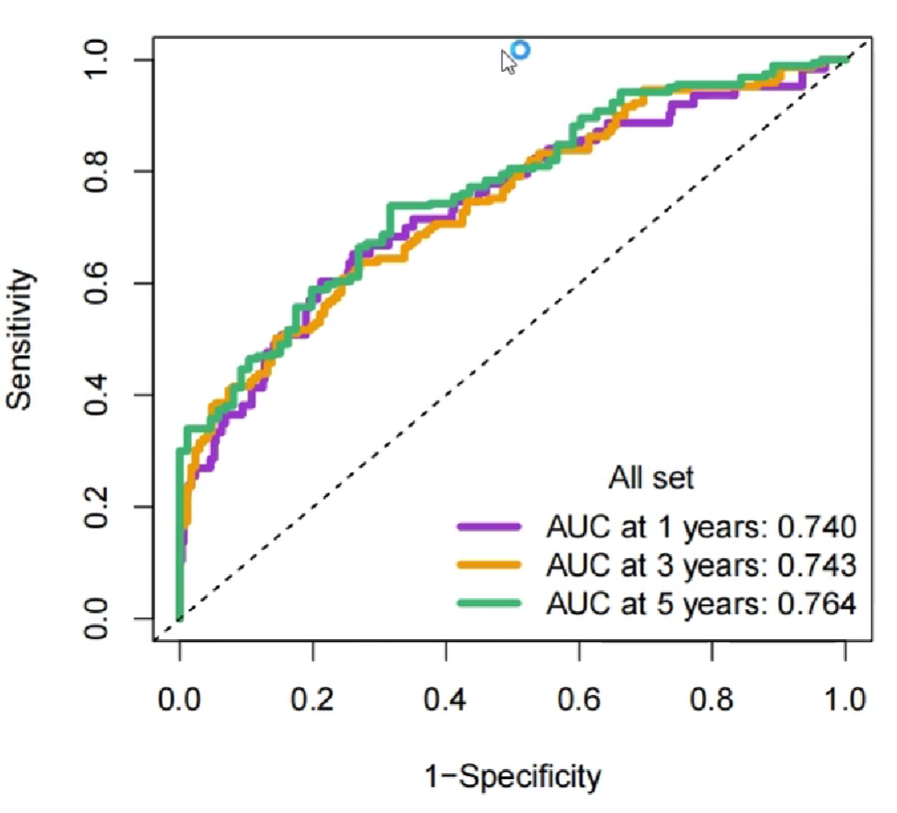
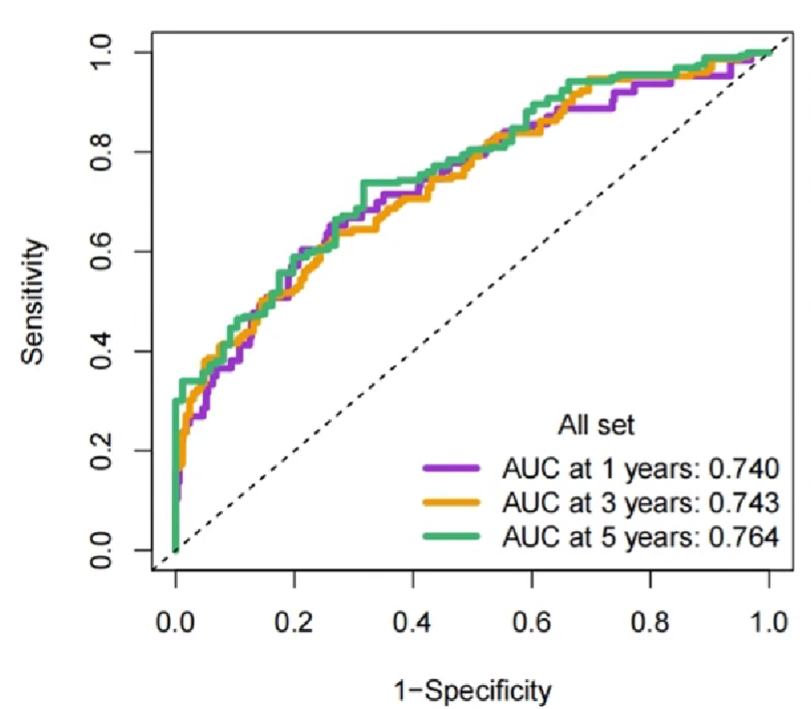
另一种ROC分析–timeROC时间依赖型生存曲线:
-
T事件时间 -
delta事件状态(删失数据值为0) -
marker一个标记值,值越大,事件越可能发生,此处使用风险得分。如果使用的数据值越小越可能发生,则可以在前面加负号 -
other_markers协变量(矩阵形式输入) -
cause所关注的时间结局,一般为1(死亡) -
weighting计算方法,默认使用KM模型,还可以是”cox”cox模型、”aalen”additive Aalen模型 -
times想计算ROC曲线的时间节点 -
ROC是否保存sensitivities的specificties值(默认为T) -
iid是否保存置信区间(默认为F,样本量大时很耗时间)
roc_rt <- timeROC(
T = risk$time,
delta = risk$state,
marker = risk$riskScore,
cause = 1,
weighting = 'aalen',
times = c(1, 3, 5),
ROC = T
);
pdf(file = "save_data\\ROC.all.pdf", width = 5, height = 5);
plot(roc_rt, time = 1, col = bioCol[1], title = F, lwd = 4);
plot(roc_rt, time = 3, col = bioCol[2], title = F, lwd = 4, add = T); # 在前一条线上继续添加
plot(roc_rt, time = 5, col = bioCol[3], title = F, lwd = 4, add = T); # 在前一条线上继续添加
legend(
'bottomright',
c(paste0('AUC at 1 year: ', sprintf("%.03f", roc_rt$AUC[1])),
paste0('AUC at 3 years: ', sprintf("%.03f", roc_rt$AUC[2])),
paste0('AUC at 5 years: ', sprintf("%.03f", roc_rt$AUC[3]))),
col = bioCol[1:3],
lwd = 4,
bty = 'n',
title = "All set"
);
dev.off();
其它临床特征的ROC曲线:
pre_time <- 5; # 预测年限
# 先使用风险得分作roc预测
roc_rt <- timeROC(
T = risk$time,
delta = risk$state,
marker = risk$riskScore,
cause = 1,
weighting = 'aalen',
times = c(pre_time),
ROC = T
);
pdf(file = "save_data\\cliROC.all.pdf", width = 5.5, height = 5.5);
# 风险得分的roc曲线
plot(roc_rt, time = pre_time, col = bioCol[1], title = F, lwd = 4);
#
abline(0, 1);
auc_text <- c(paste0("Risk", ", AUC=", sprintf("%.3f", roc_rt$AUC[2])));
# 再使用临床数据的其它列作roc预测
for (i in 4:ncol(rt)) {
roc_rt <- timeROC(
T = rt$time,
delta = rt$state,
marker = rt[, i],
cause = 1,
weighting = 'aalen',
times = c(pre_time),
ROC = T
);
plot(roc_rt, time = pre_time, col = bioCol[i-2], title = F, lwd = 4, add = T);
auc_text <- c(auc_text, paste0(colnames(rt)[i], ", AUC=", sprintf("%.3f", roc_rt$AUC[2])));
}
legend("bottomright", auc_text, lwd = 4, bty = 'n', title = "All set", col = bioCol[1:(ncol(rt)-1)]);
dev.off();
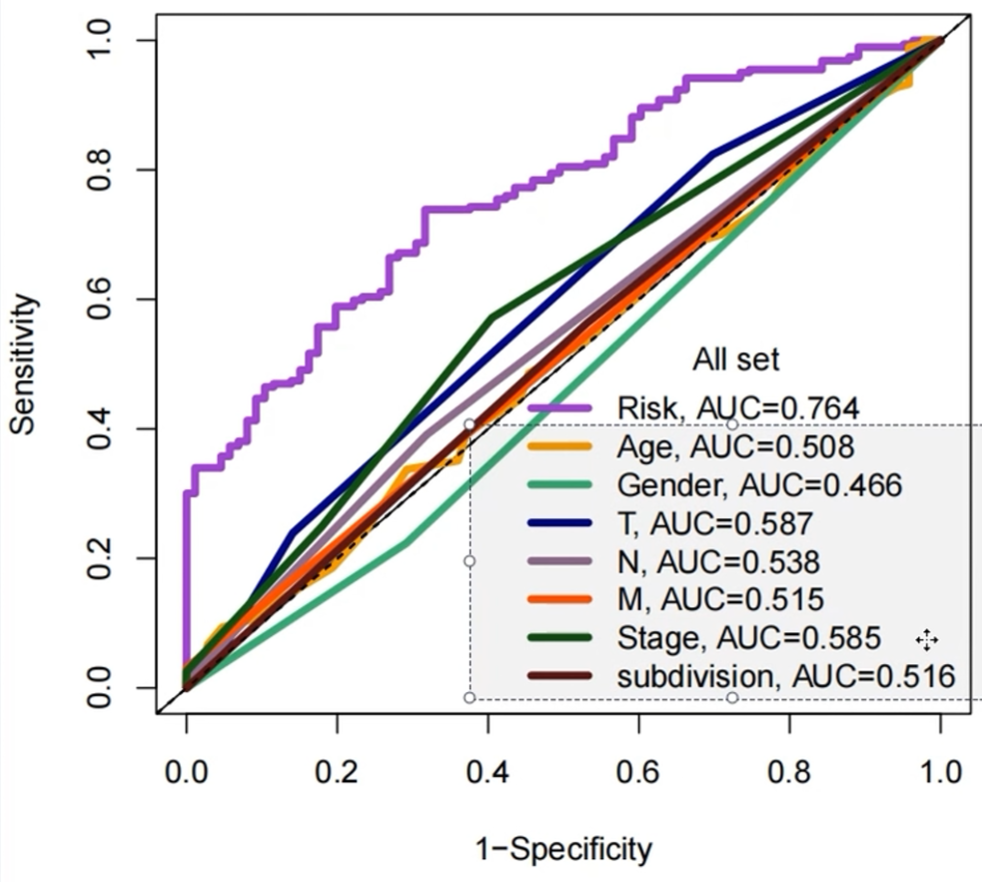
可以看到使用风险得分进行roc预测的准确度明显大于其它临床特征
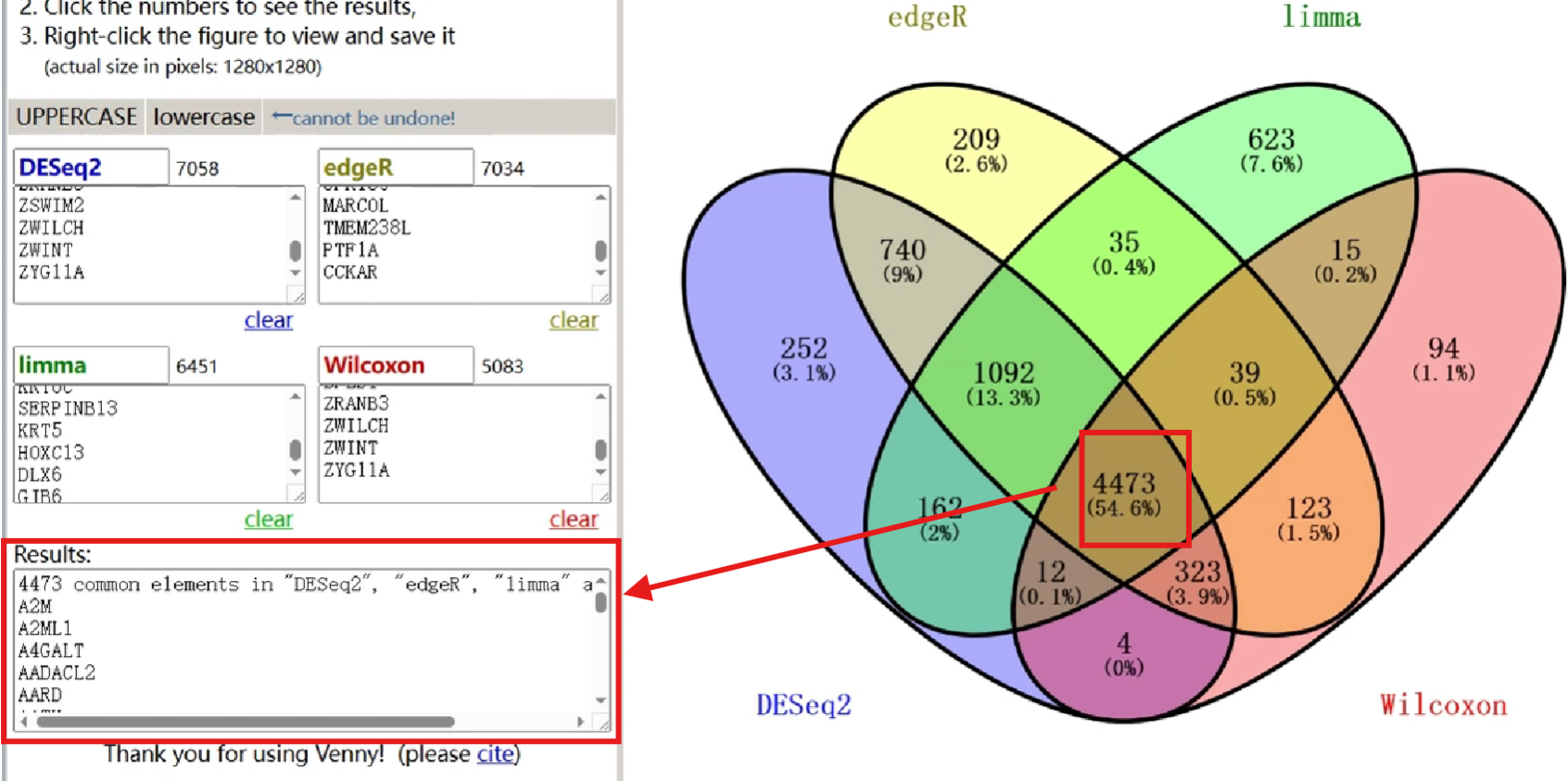
Venn图绘制
除了使用代码绘制,还可以使用在线网站
将4种差异表达分析结果基因输入,并调整相关设置
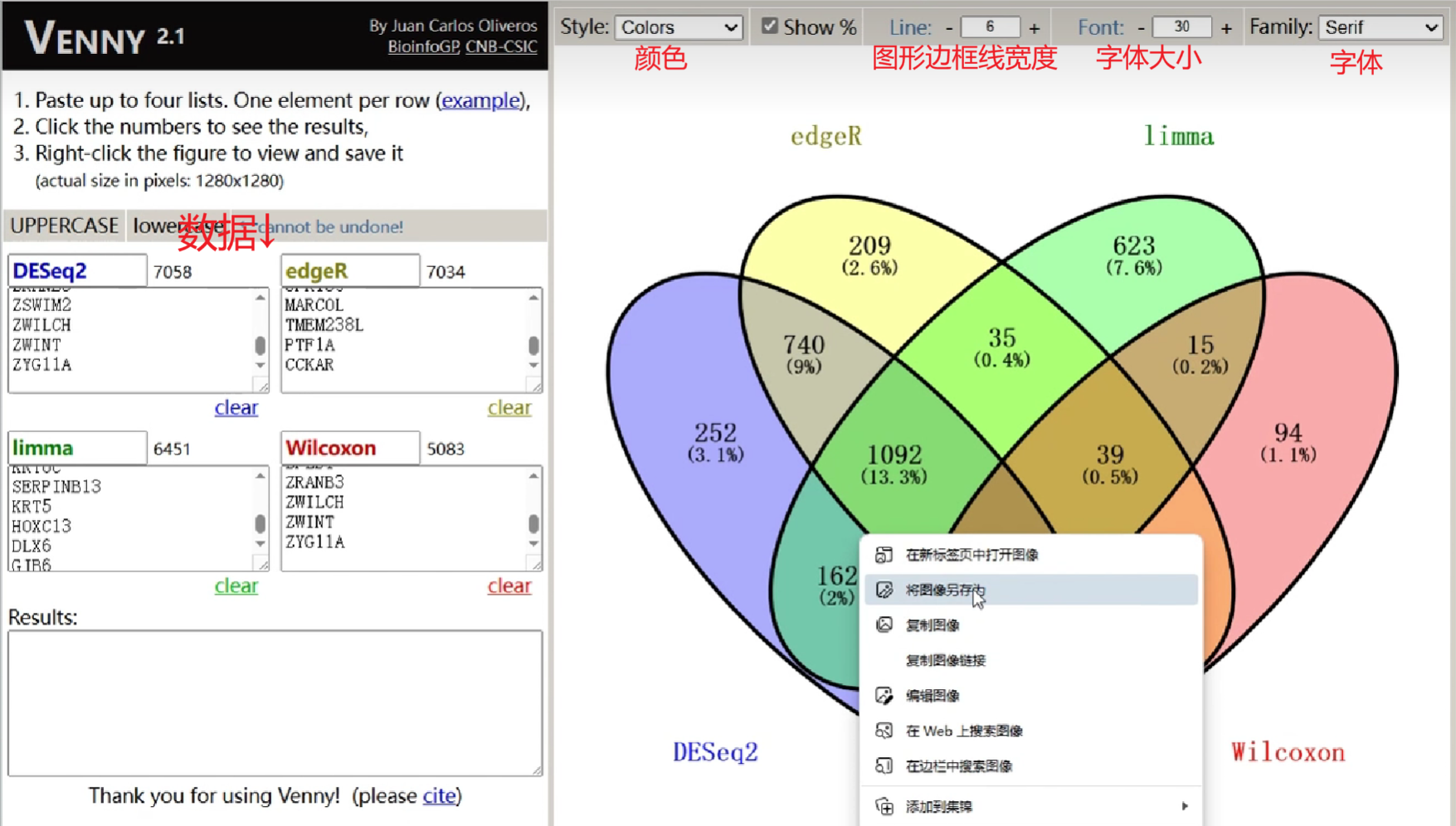
之后右键图片->将图像另存为
除此之外,点击图片中的各区域,左下角results就可列出这部分交叉的基因
列线图
使用regplot包的版本为0.2,下载
library(survival);
if(!require("regplot", quietly = T))
{
install.packages("regplot");
library("regplot");
}
library(rms);
读取风险得分和临床数据,合并:
# 风险得分
risk <- read.table("save_data\\risk.txt", check.names = F, row.names = 1, sep = '\t', header = T);
# 临床数据
library("readxl");
library(tibble);
cli <- read_excel("save_data\\new_clinical.xlsx");
cli <- column_to_rownames(cli, "ID");
# 删除包含NA的行
cli <- cli[apply(cli, 1, function(x)any(is.na(match(NA, x)))), ];
# 合并
same_sample <- intersect(rownames(risk), rownames(cli)); # 共同样本名
risk <- risk[same_sample, ];
cli <- cli[same_sample, ]; # 过滤
rt <- cbind(risk[, c("time", "state", "risk")], cli[, c("Age", "Gender", "T", "N", "M", "Stage", "subdivision")]); # 合并
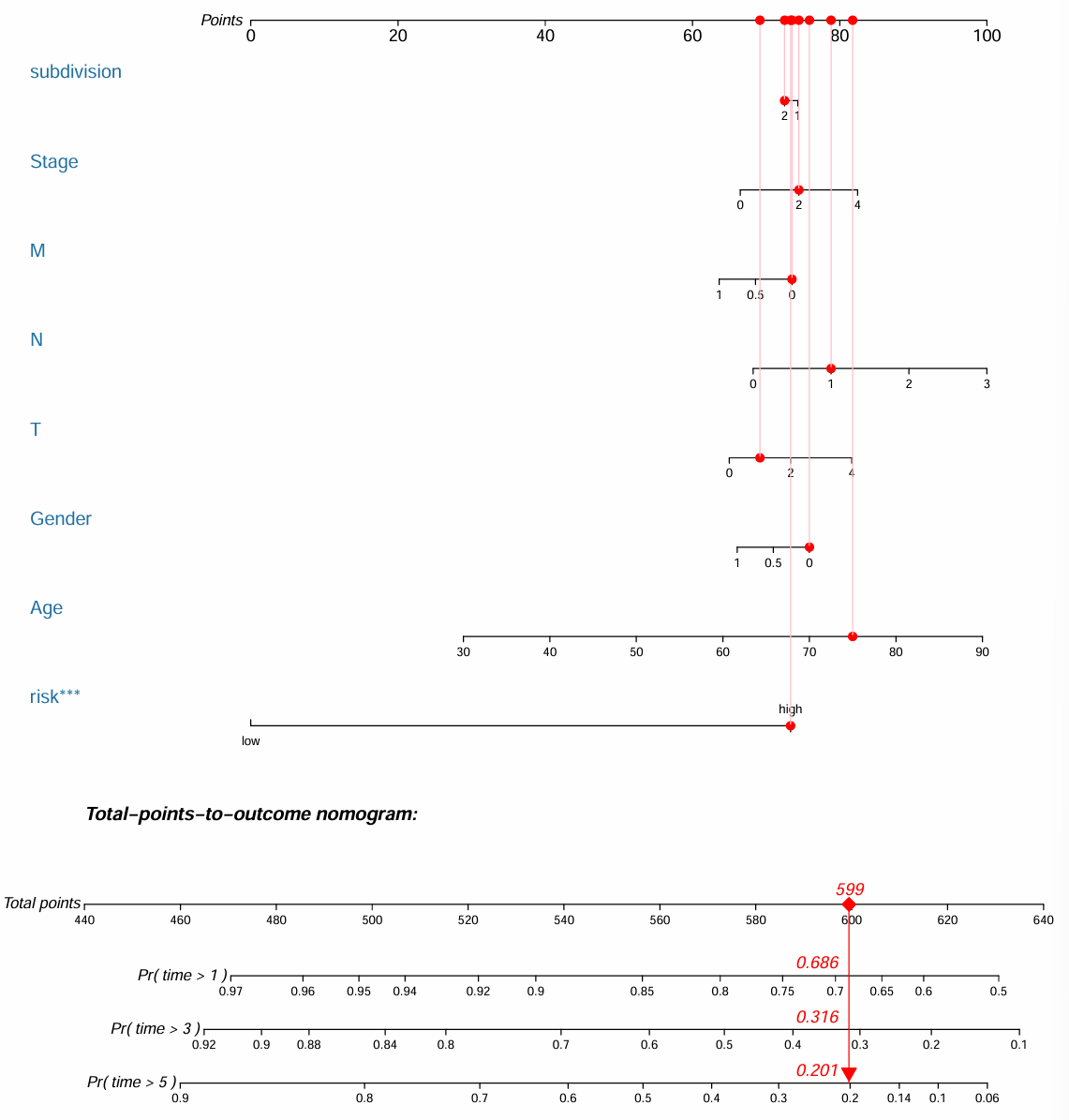
画列线图:
# 因为我们这里有生存时间和生存状态,所以用cox
# 如果只是用来预测二分类(如正常/肿瘤),就用logistic回归
res.cox <- coxph(Surv(time, state) ~ ., data = rt);
pdf(file = "save_data\\nomogram.pdf", width = 10, height = 11);
# 画图
nom1 <- regplot(
res.cox,
title = "",
points = T,
droplines = T,
observation = rt[50, ],
failtime = c(1, 3, 5),
prfail = F
);
dev.off();
更高版本的regplot包画出的列线图:
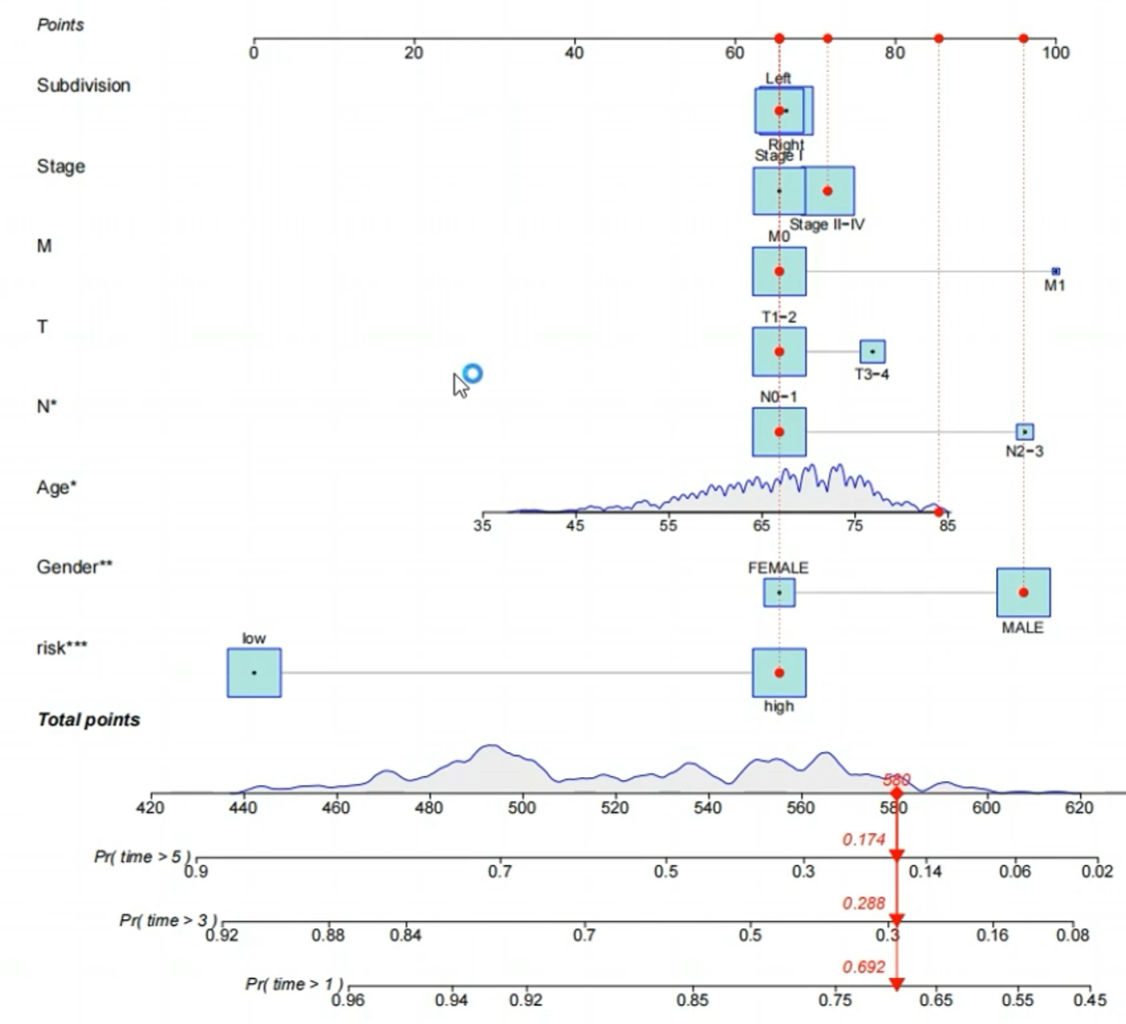
图左侧标明了画图过程中使用的变量,图右侧的points区域是每个变量相应能获得多少分,根据所有变量分数之和可以获得总体的打分Total Points,这个总体打分可以预测每个样本存活1/3/5年的概率
列线图打分:
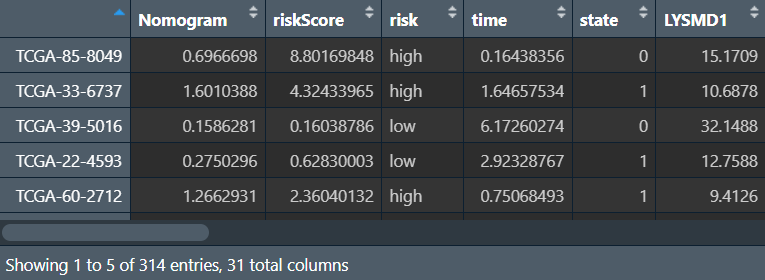
nomo_risk <- predict(res.cox, data = rt, type = "risk");
rt <- cbind(Nomogram = nomo_risk, risk);
outTab <- rbind(ID = colnames(rt), rt);
write.table(outTab, file = "save_data\\nomoRisk.txt", sep = '\t', row.names = F, quote = F);
校准曲线:
pdf(file = "save_data\\calibration.pdf", width = 6, height = 6);
# 1年
f <- cph(Surv(time, state) ~ Nomogram, x = T, y = T, surv = T, data = rt, time.inc = 1);
cal <- calibrate(f, cmethod = 'KM', method = "boot", u = 1, m = (nrow(rt)/3), B = 1000);
plot(
cal,
xlim = c(0, 1),
ylim = c(0, 1),
xlab = "Nomogram-predicted OS (%)",
ylab = "Observed OS (%)",
lwd = 3,
col = "Firebrick2",
sub = F
);
# 3年
f <- cph(Surv(time, state) ~ Nomogram, x = T, y = T, surv = T, data = rt, time.inc = 3);
cal <- calibrate(f, cmethod = 'KM', method = "boot", u = 3, m = (nrow(rt)/3), B = 1000);
plot(
cal,
xlim = c(0, 1),
ylim = c(0, 1),
xlab = "Nomogram-predicted OS (%)",
ylab = "Observed OS (%)",
lwd = 3,
col = "MediumSeaGreen",
sub = F,
add = T
);
# 5年
f <- cph(Surv(time, state) ~ Nomogram, x = T, y = T, surv = T, data = rt, time.inc = 5);
cal <- calibrate(f, cmethod = 'KM', method = "boot", u = 5, m = (nrow(rt)/3), B = 1000);
plot(
cal,
xlim = c(0, 1),
ylim = c(0, 1),
xlab = "Nomogram-predicted OS (%)",
ylab = "Observed OS (%)",
lwd = 3,
col = "NavyBlue",
sub = F,
add = T
);
# 图例
legend(
"bottomright",
c("1-year", "3-year", "5-year"),
col = c("Firebrick2", "MediumSeaGreen", "NavyBlue"),
lwd = 3,
bty = 'n'
);
dev.off();
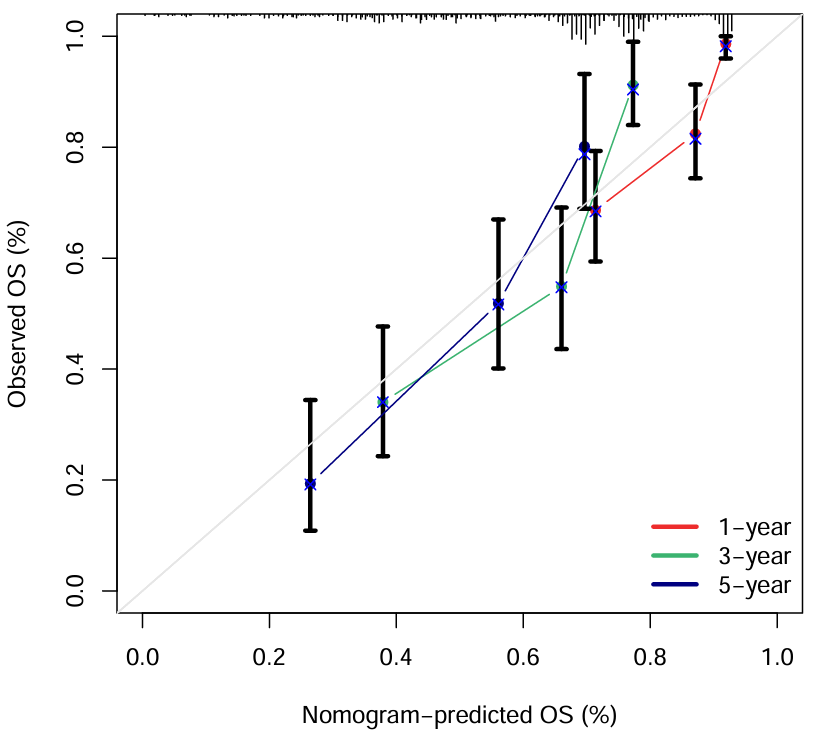
横坐标是这个列线图预测的总体生存率,纵坐标是实际生存率。当预测==实际(即曲线趋近对角线)时,预测效果较好
免疫组化图片
使用HPA数据库,它是基于蛋白质组学、转录组学以及系统生物学数据创建的,拥有组织、细胞、器官等图谱,不仅收录肿瘤组织,也涵盖正常组织的蛋白表达情况,还可查询肿瘤患者的生存曲线
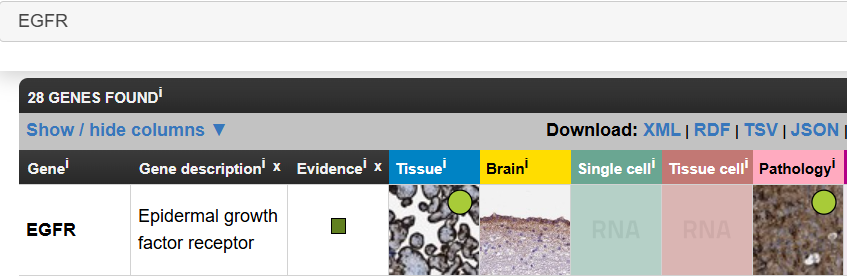
以EGFR基因为例:进入HPA官网,在搜索栏中输入EGFR,search
-
Tissue是正常组织 -
Pathology是肿瘤组织
-
RNA expression (nTPM)是RNA表达水平 -
Protein expression (score)是蛋白表达水平
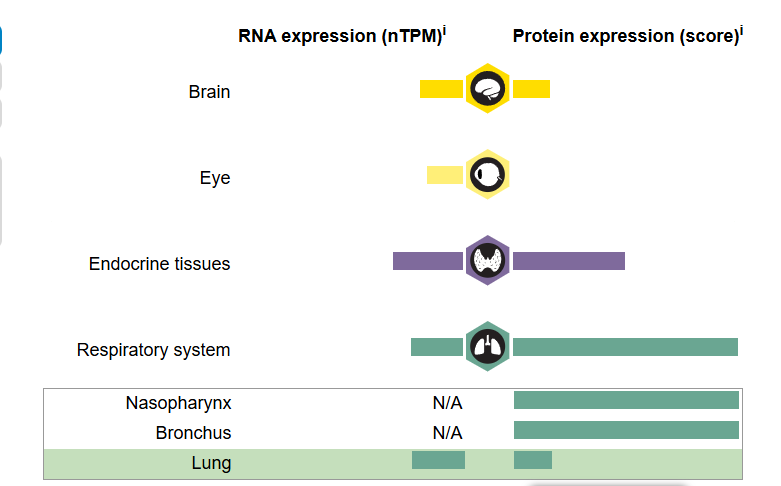
点击Respiratory system->Lung,就可以查看该基因在肺中的表达
Antibody HPA001200、Antibody HPA018530、…这些都是抗体名称
以HPA018530为例,点击对应的图片
{kind=link}
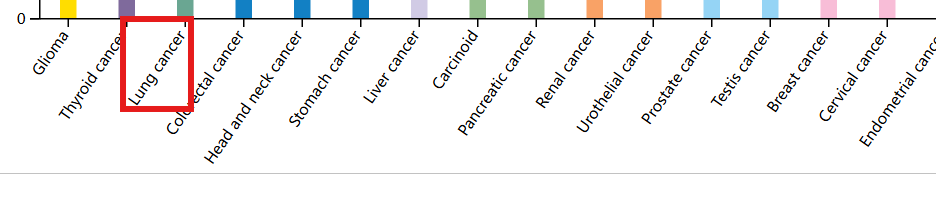
对于肿瘤组织,点击页面下方的Lung cancer就可查看该基因在肺癌中的表达
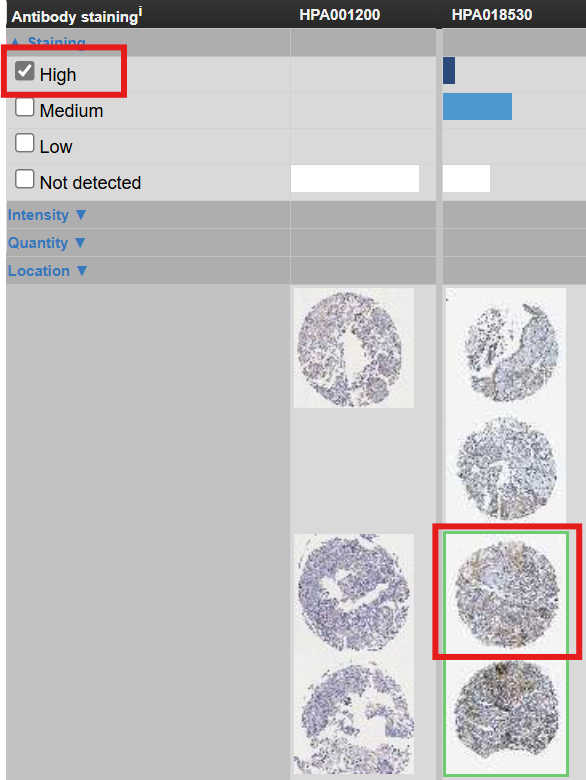
在新打开的页面中,Antibody staining标识了各抗体的免疫组化图片,点击HPA018530,勾选high(高表达),点击绿框标注的图片
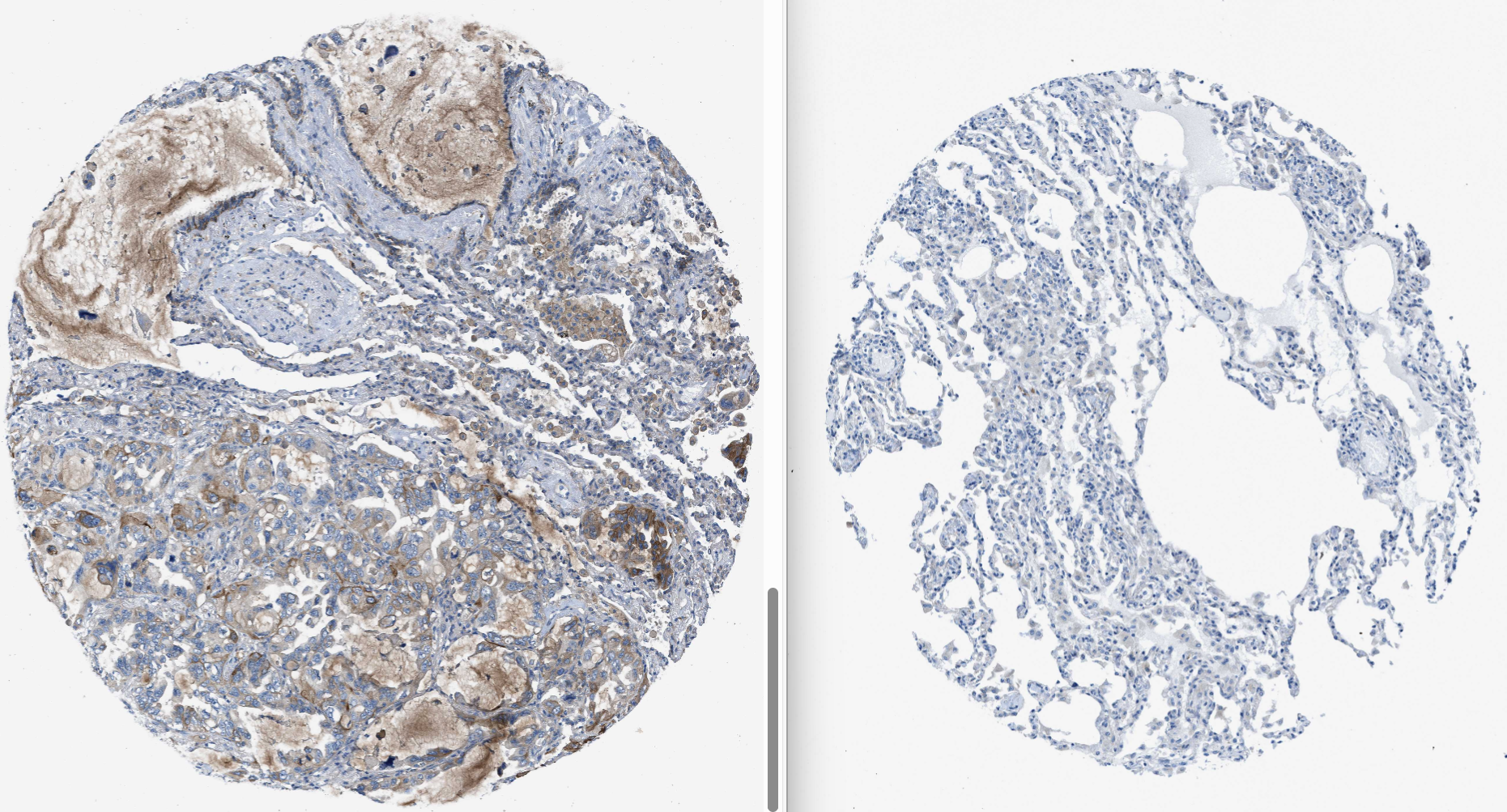
{kind=link}
对比这两种图片,就可得到该抗体在正常/肿瘤免疫组化的结果
免疫细胞浸润分析
免疫细胞浸润分析(cibersort)
需要数据:tpm表达矩阵
需要包:e1071、parallel、preprocessCore、bseqsc、tidyverse、corrplot、vioplot、CIBERSORT
if(!require("e1071", quietly = T))
{
install.packages("e1071");
}
if(!require("parallel", quietly = T))
{
install.packages("parallel");
}
if(!require("preprocessCore", quietly = T))
{
library(BiocManager);
BiocManager::install("preprocessCore");
}
if(!require("devtools", quietly = T))
{
install.packages("devtools");
}
if(!require("corrplot", quietly = T))
{
install.packages("corrplot");
}
if(!require("vioplot", quietly = T))
{
install.packages("vioplot");
}
if(!require("bseqsc", quietly = T))
{
library(devtools);
devtools::install_github("shenorrlab/bseqsc");
}
if(!require("CIBERSORT", quietly = T))
{
library(devtools);
devtools::install_github("Moonerss/CIBERSORT");
}
library(devtools);
library(e1071);
library(preprocessCore);
library(parallel);
library(bseqsc);
library(ggplot2);
library(CIBERSORT);
library(corrplot);
library(vioplot);
读取数据:
data(LM22); # 导入CIBERSORT内置数据,包含22种细胞中基因表达情况
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", check.names = F, row.names = 1, sep = '\t', header = T);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
进行CIBERSORT分析:
res <- cibersort(sig_matrix = LM22, mixture_file = data);
# 保存结果
res <- as.matrix(res[, 1:(ncol(res)-3)]);
res_save <- cbind(id = rownames(res), res);
write.table(res_save, file = "save_data\\CIBERSORT-Results.txt", row.names = F, sep = '\t', quote = F);
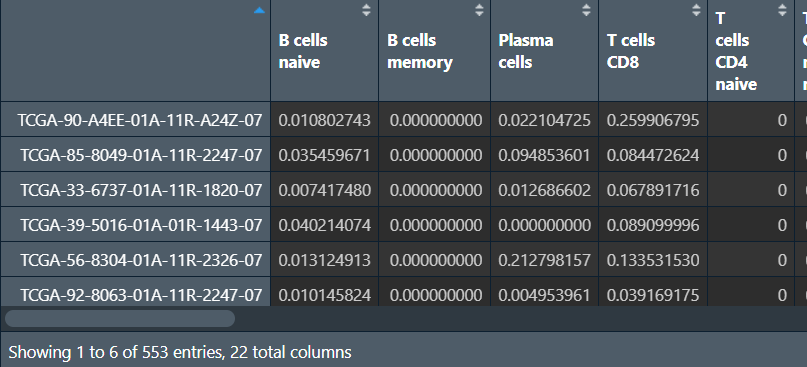
行名是样本名,列名是不同的细胞类型,后三列是p值、相关性、RMSE,这三列一般不用,删去
绘图–柱状图1:
immune <- read.table("save_data\\CIBERSORT-Results.txt", header = T, sep = '\t', check.names = F);
library(tidyverse);
immune <- column_to_rownames(immune, "id");
immune <- as.matrix(immune);
data <- t(immune);
col <- rainbow(nrow(data), s = 0.7, v = 0.7);
pdf(file = "save_data\\CIBERSORT1.pdf", width = 22, height = 10);
par(las = 1, mar = c(8, 5, 4, 16), mgp = c(3, 0.1, 0), cex.axis = 1.5);
a1 <- barplot(data, col = col, yaxt = "n", ylab = "Relative Percent", xaxt = "n", cex.lab = 1.8);
a2 <- axis(2, tick = F, labels = F);
axis(2, a2, paste0(a2*100, "%"));
axis(1, a1, labels = F);
par(srt = 60, xpd = T);
text(a1, -0.02, colnames(data), adj = 1, cex = 0.6);
par(srt = 0);
ytick2 <- cumsum(data[, ncol(data)]);
ytick1 <- c(0, ytick2[-length(ytick2)]);
legend(
par("usr")[2]*0.98,
par("usr")[4],
legend = rownames(data),
col = col, pch = 15, bty = "n", cex = 1.3
);
dev.off();
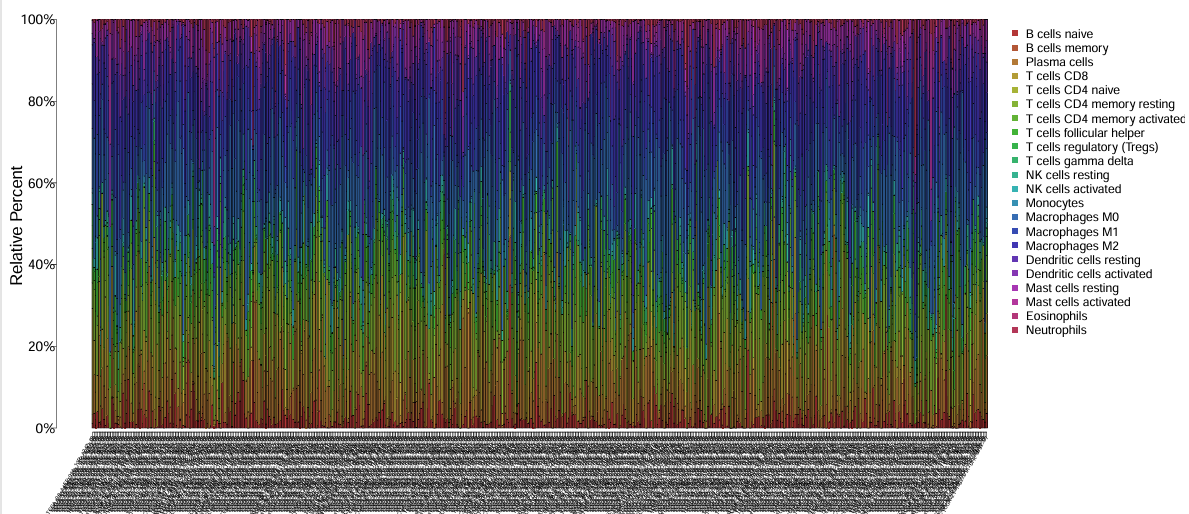
颜色代表细胞类型,横坐标是不同的样本,纵坐标是不同类型的细胞在不同样本中所占的比例
第二种柱状图:
# 数据准备
cell.prop <- apply(immune, 1, function(x){x/sum(x)});
plot_data <- data.frame();
for(i in 1:ncol(cell.prop)){
plot_data <- rbind(
plot_data,
cbind(
cell.prop[, i],
rownames(cell.prop),
rep(colnames(cell.prop)[i], nrow(cell.prop))
)
);
}
colnames(plot_data) <- c("proportion", "celltype", "sample");
plot_data$proportion <- as.numeric(plot_data$proportion);
# 绘图
my_colors36 <- c('#E5D2DD','#53A85F','#F1BB72','#F3B1A0','#D6E7A3','#57C3F3','#476D87','#E95C59','#E59CC4','#AB3282','#23452F','#BD956A','#8C549C','#585658','#9FA3A8','#E0D4CA','#5F3D69','#C5DEBA','#58A4C3','#E4C755','#F7F398','#AA9A59','#E63863','#E39A35','#C1E6F3','#6778AE','#91D0BE','#B53E2B','#712820','#DCC1DD','#CCE0F5','#CCC9E6','#625D9E','#68A180','#3A6963','#968175');
pdf(file = "save_data\\CIBERSORT2.pdf", width = 22, height = 10);
ggplot(
plot_data,
aes(sample, proportion, fill = celltype),
) +
geom_bar(stat = "identity", position = "fill") +
scale_fill_manual(values = my_colors36) +
ggtitle("cell portation") +
theme_bw() +
theme(
axis.ticks.length = unit(0.5, "cm"),
axis.title.x = element_text(size = 1),
axis.text.x = element_text(angle = 45, hjust = 0.5, vjust = 0.5)
) +
guides(fill = guide_legend(title = NULL));
dev.off();
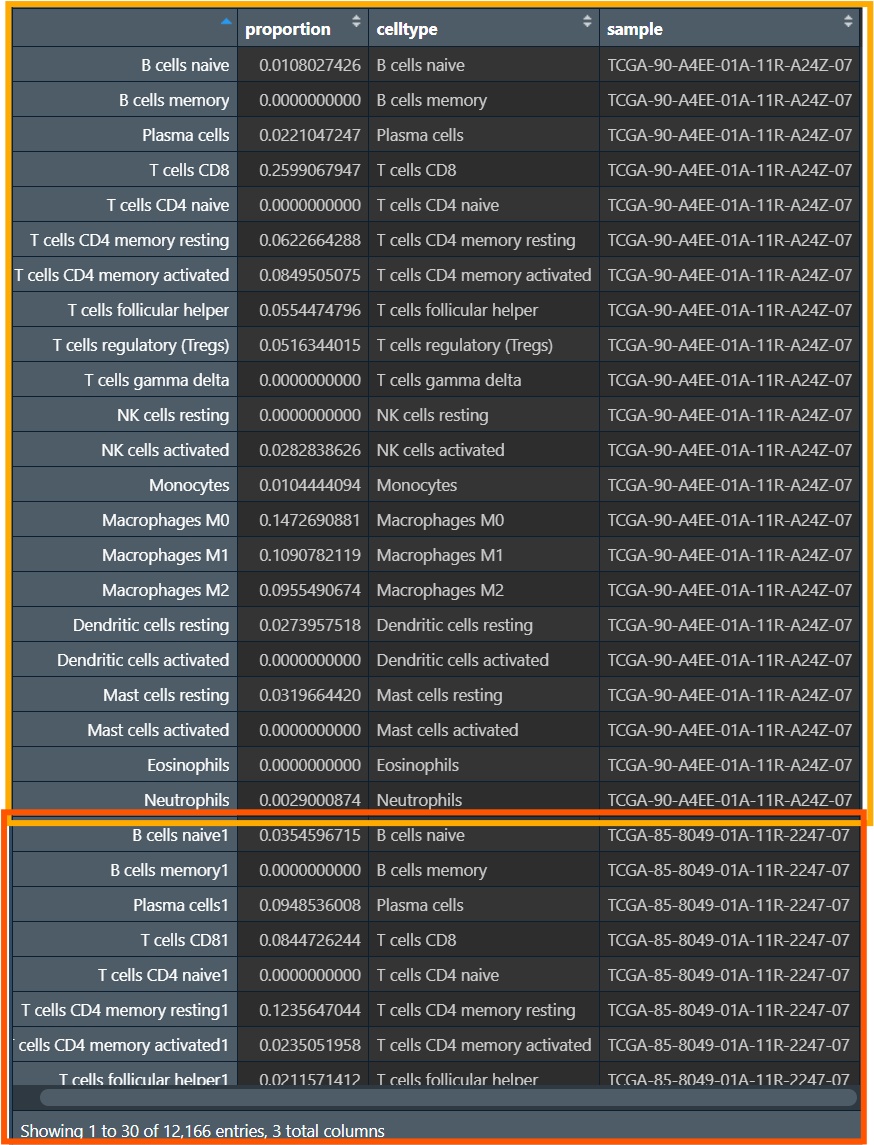
宽数据->长数据:将每个样本的每种细胞占比(数据值)变为proportion列
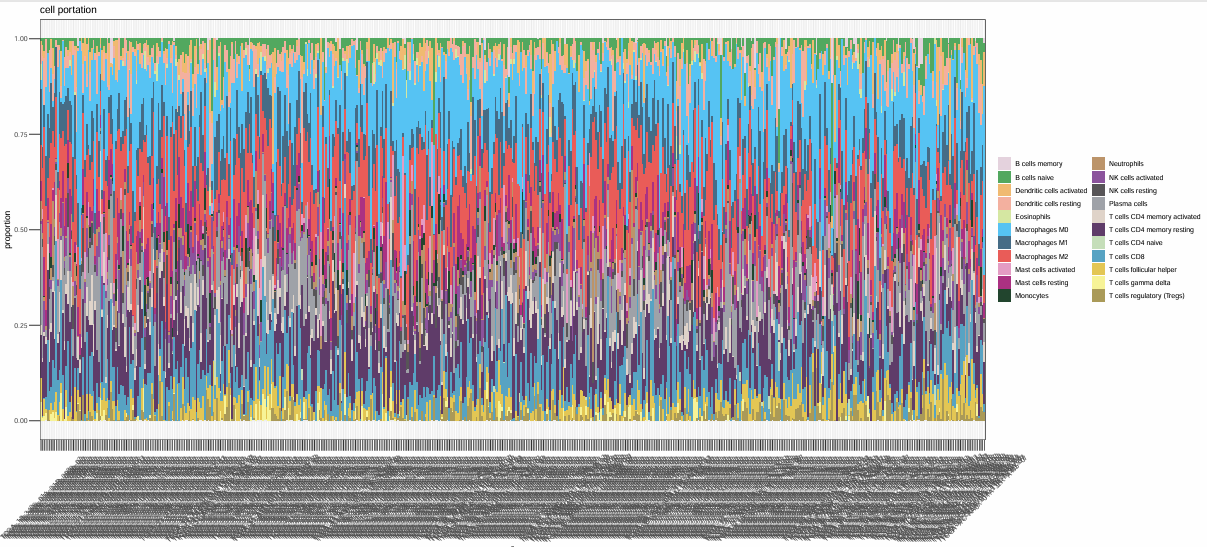
相关性图:
pdf(file = "save_data\\CIBERSORT_cor.pdf", width = 13, height = 13);
par(oma = c(0.5, 1, 1, 1.2));
plot_data <- immune[, colMeans(immune)>0];
plot_data <- cor(immune);
corrplot(
plot_data,
order = "hclust",
method = "color",
addCoef.col = "black",
diag = T,
tl.col = "black",
col = colorRampPalette(c("blue", "white", "red"))(50)
);
dev.off();
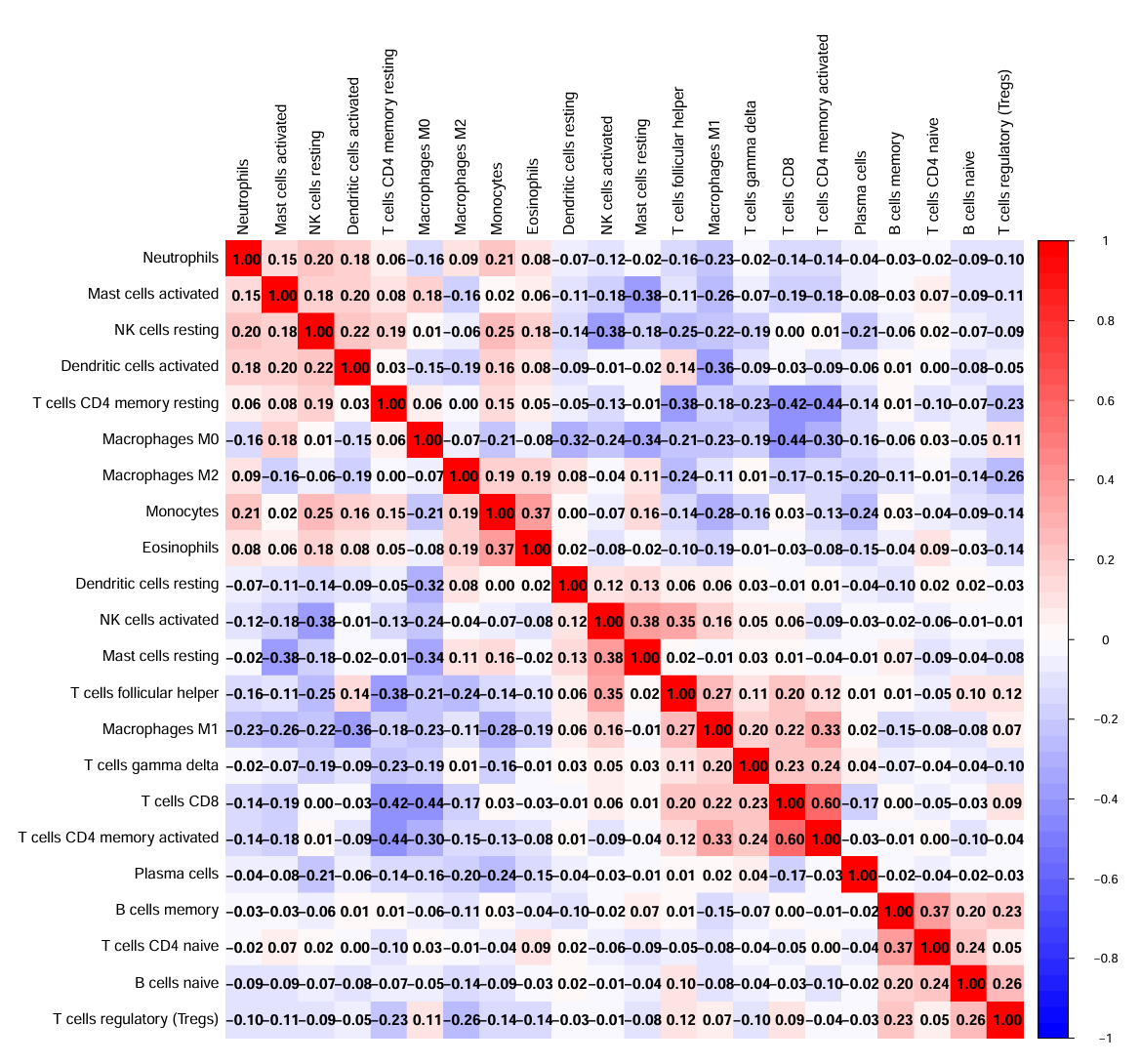
表示这22种细胞类型之间,红色–正相关、蓝色–负相关、白色–不相关
对正常组和肿瘤组的免疫细胞浸润进行差异分析:
# 先根据样本名分成正常组和肿瘤组(同之前操作)
group <- sapply(strsplit(rownames(immune), '\\-'), "[", 4);
group <- sapply(strsplit(group, ''), "[", 1);
group <- gsub("2", "1", group);
con_num <- length(group[group==1]);
treat_num <- length(group[group==0]);
type <- c(rep(1, con_num), rep(2, treat_num));
rt1 <- immune[group==1, ];
rt2 <- immune[group==0, ];
rt <- rbind(rt1, rt2);
# 绘制小提琴图
outTab <- data.frame();
pdf(file = "save_data\\CIBERSORT_vioplot.pdf", width = 13, height = 8);
par(las = 1, mar = c(10, 6, 3, 3));
x <- 1:ncol(rt);
y <- 1:ncol(rt);
plot(
x, y,
xlim = c(0, 63),
ylim = c(min(rt), max(rt)+0.05),
main = "",
xlab = "",
ylab = "Fraction",
pch = 21,
col = "white",
xaxt = "n"
);
for(i in 1:ncol(rt)){
# 计算标准差
if(sd(rt[1:con_num])==0){
rt[1, i] <- 0.00001;
}
if(sd(rt[(con_num+1):(con_num+treat_num), i])==0){
rt[con_num+1, i] <- 0.00001;
}
# 取出数据
rt1 <- rt[1:con_num, i];
rt2 <- rt[(con_num+1):(con_num+treat_num), i];
# 绘图
vioplot(rt1, at = 3*(i-1), lty = 1, add = T, col = "blue");
vioplot(rt2, at = 3*(i-1)+1, lty = 1, add = T, col = "red");
# Wilcox检验
wilcox_test <- wilcox.test(rt1, rt2);
p <- wilcox_test$p.value;
if(p<0.05){
cell_p <- cbind(Cell=colnames(rt)[i], pvalue = p);
outTab <- rbind(outTab, cell_p);
}
mx <- max(c(rt1, rt2));
lines(c(x=3*(i-1)+0.2, x=3*(i-1)+0.8), c(mx, mx));
text(
x = 3*(i-1)+0.5,
y = mx+0.02,
labels = ifelse(
p<0.001,
paste0("p<0.001"),
paste0("p=", sprintf("%.03f", p))
)
);
}
legend(
"topright",
c("Normal", "Tumor"),
lwd = 3,
bty = "n",
cex = 1,
col = c("blue", "red")
);
text(
seq(1, 64, 3),
-0.04,
xpd = NA,
labels = colnames(rt),
cex = 1,
srt = 45,
pos = 2
);
dev.off();
# 保存数据(免疫细胞和其p值)
write.table(outTab, file = "save_data\\CIBERSORT_Diff.txt", row.names = F, sep = '\t', quote = F);
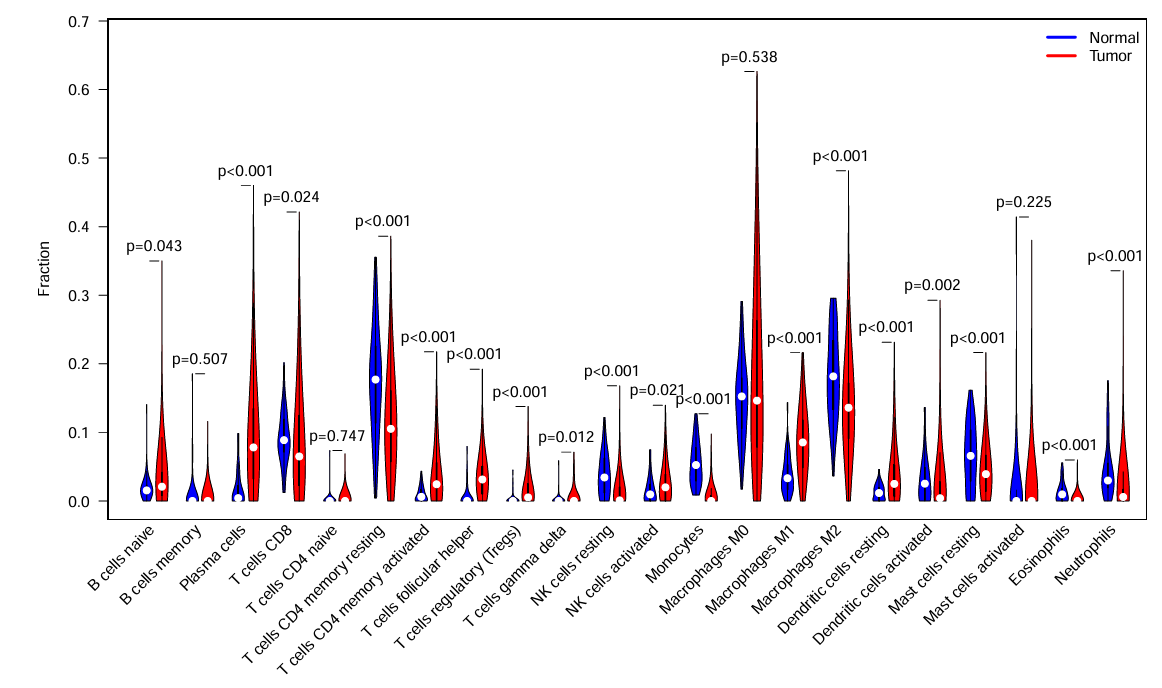
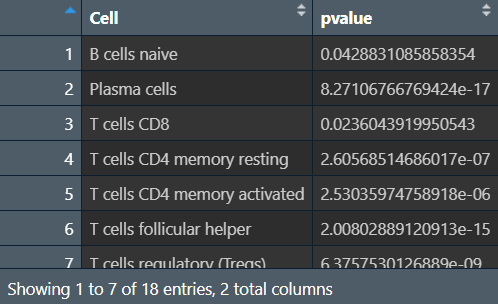
横坐标是细胞种类,纵坐标是每种细胞具体含量,p值就是每种细胞在正常/肿瘤组中的差异
outTab是在正常/肿瘤组中有统计学差异的细胞
除此之外,正常/肿瘤组也可以换成高/低风险组、某基因表达量高/低等
多种免疫细胞浸润分析
library(limma);
library(scales);
library(ggplot2);
library(ggtext);
读取免疫细胞浸润文件、tpm表达矩阵,并取交集:
# 免疫细胞浸润文件
immune <- read.csv("data\\infiltration_estimation_for_tcga.csv", check.names = F, row.names = 1, sep = ',', header = T);
immune <- as.matrix(immune);
rownames(immune) <- gsub( # 改样本名
"(.*?)\\-(.*?)\\-(.*?)\\-(.*)",
"\\1\\-\\2\\-\\3",
rownames(immune)
);
immune <- avereps(immune); # 对相同样本取平均值
# tpm表达矩阵
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", check.names = F, row.names = 1, sep = '\t', header = T);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
group <- sapply(strsplit(colnames(data),"\\-"), "[", 4);
group <- sapply(strsplit(group,""), "[", 1);
data <- data[,group == 0]; # 仅保留肿瘤样本
data <- t(data);
rownames(data) <- substr(rownames(data), 1, 12);
rownames(data) <- gsub('[.]', '-', rownames(data));
# 取交集
same_sample <- intersect(row.names(data), row.names(immune));
# 以A1BG这个基因的表达水平为区分
data <- data[same_sample, "A1BG"];
immune <- immune[same_sample, ];
data:
是A1BG基因在各样本中的表达量
免疫细胞浸润文件immune:
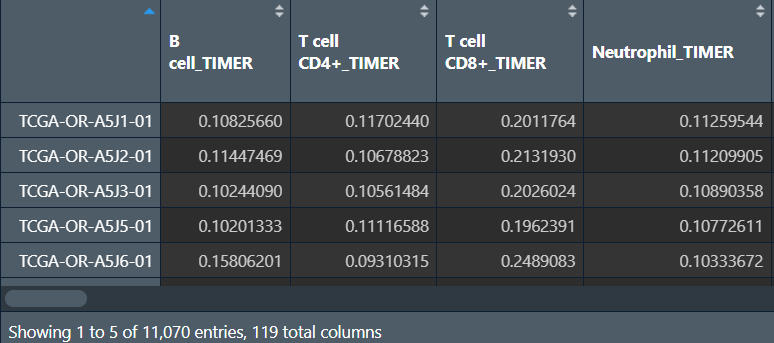
包括所有TCGA样本、使用不同分析方法进行分析的、各种免疫细胞分析结果
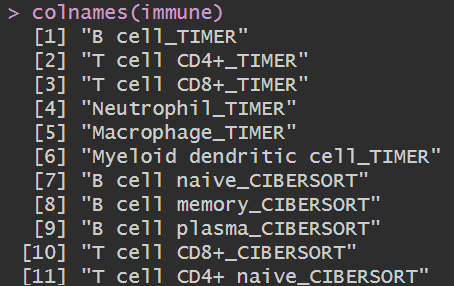
列名后面的TIMER、CIBERSORT就是分析方法
此时得到的immune与上节中的类似,只不过它是多种分析方法得到的结果,可根据列名选取某种分析方法得到的结果,绘图的方法与上节课相同(此处画了气泡图)
相关性分析(A1BG基因表达量与免疫细胞分析结果的相关性):
x <- as.numeric(data);
outTab <- data.frame();
for(i in colnames(immune)){
y <- as.numeric(immune[, i]);
corT <- cor.test(x, y, method="spearman");
cor <- corT$estimate;
pvalue <- corT$p.value;
if(pvalue<0.05){
outTab <- rbind(outTab, cbind(immune=i, cor, pvalue));
}
}
# 保存结果
write.table(file="save_data\\immune_corResult.txt", outTab, sep="\t", quote=F, row.names=F);
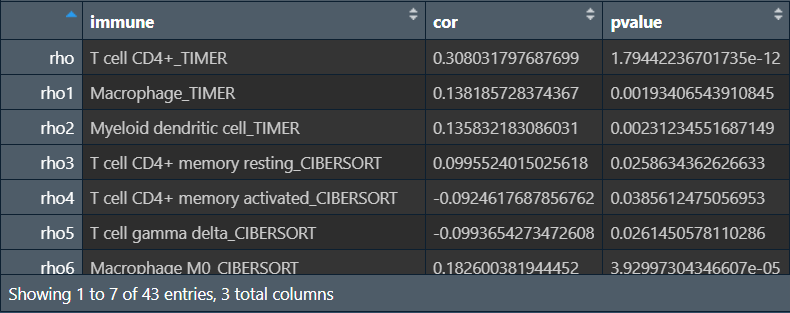
得到与A1BG基因表达量相关性显著的免疫细胞
绘制气泡图:
corResult <- read.table("save_data\\immune_corResult.txt", head=T, sep="\t");
corResult$Software <- sapply(strsplit(corResult[, 1], "_"), '[', 2); # 得到分析方法Software列
corResult$Software <- factor(
corResult$Software,
level = as.character(
unique(
corResult$Software[rev(order(as.character(corResult$Software)))]
))); # 将Software列转为factor
b <- corResult[order(corResult$Software), ]; # 按分析方法重排序
b$immune <- factor(b$immune, levels = rev(as.character(b$immune))); # 将immune列转为factor(如果不转,y轴将不会按分析方法排序)
colslabels <- rep(
hue_pal()(length(levels(b$Software))),
table(b$Software)
); # 绘图颜色
pdf(file = "save_data\\immune_cor.pdf", width = 10, height = 10);
ggplot(
data = b,
aes(x=cor, y=immune, color=Software)
) +
labs(x = "Correlation coefficient", y = "Immune cell") +
geom_point(size = 4.1) +
theme(
panel.background = element_rect(fill = "white", size = 1, color = "black"),
panel.grid = element_line(color = "grey75", size = 0.5),
axis.ticks = element_line(size = 0.5),
axis.text.y = ggtext::element_markdown(colour = rev(colslabels))
);
dev.off();
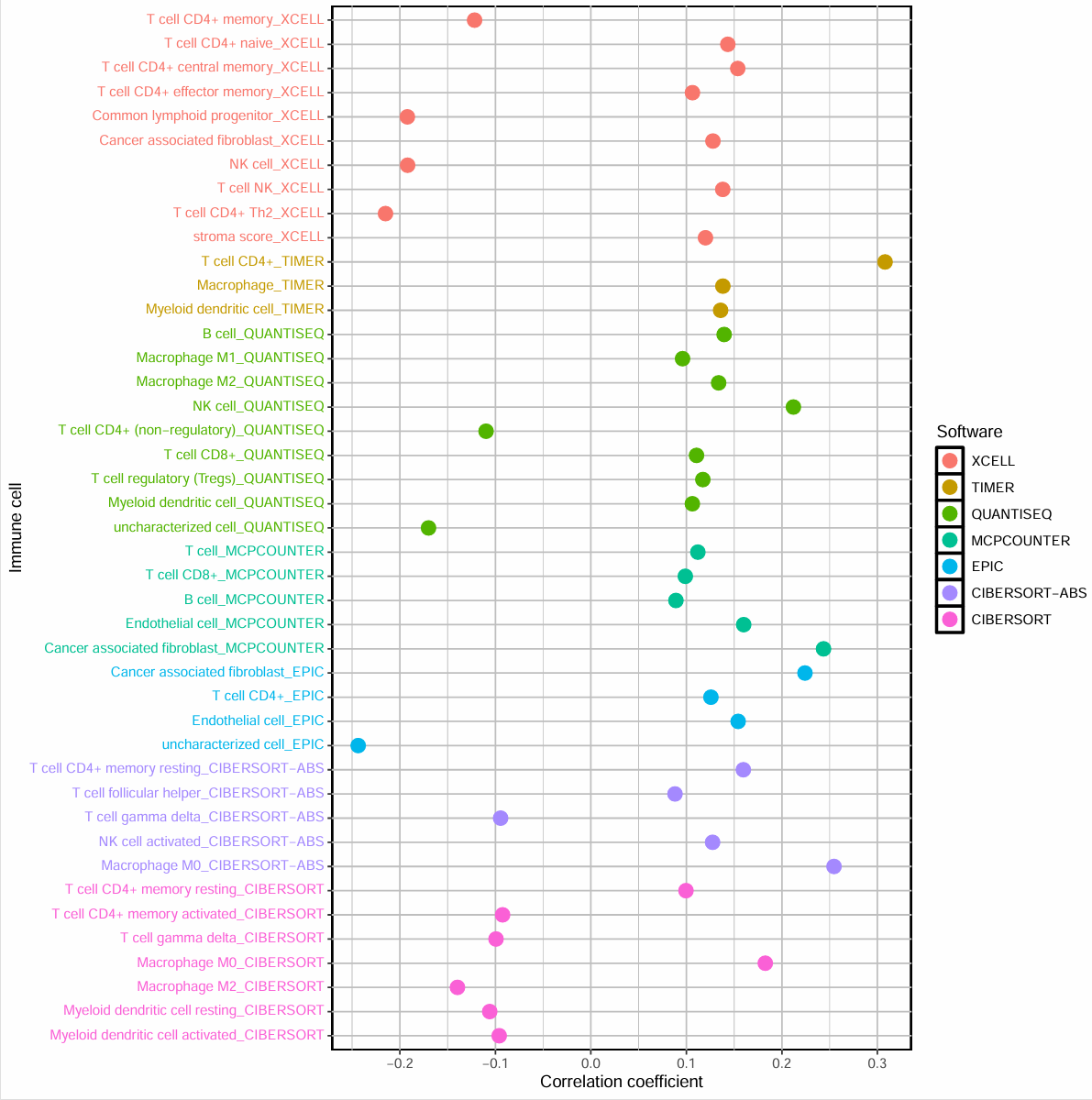
横坐标是相关性系数,每种颜色是不同的分析方法,纵坐标是不同的免疫细胞,>0的点是正相关,<0的是负相关
免疫细胞浸润分析(ssGSEA)
需要数据:tpm表达矩阵、风险得分、免疫细胞的gmt数据集(提供每种免疫细胞相关联的基因)
library(limma);
library(GSVA);
library(GSEABase);
library(ggpubr);
library(reshape2);
读取数据:
# tpm表达矩阵
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", check.names = F, row.names = 1, sep = '\t', header = T);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
# 数据集文件
geneSet <- getGmt("data\\免疫数据\\immune.gmt", geneIdType = SymbolIdentifier());
# 风险得分
risk <- read.table( "save_data\\risk.txt", check.names = F, row.names = 1, sep = '\t', header = T);
ssGSEA分析:
gsvapar <- gsvaParam(data, geneSet, kcdf = 'Gaussian', absRanking = TRUE);
ssgseaScore <- gsva(gsvapar);
normalize <- function(x){ # 标准化函数
return((x-min(x))/(max(x)-min(x)));
}
ssgseaScore <- normalize(ssgseaScore); # 对ssGSEA score进行矫正
ssgseaOut <- rbind(id = colnames(ssgseaScore), ssgseaScore);
write.table(ssgseaOut, file="save_data\\immScore.txt", sep="\t", quote=F, col.names=F);
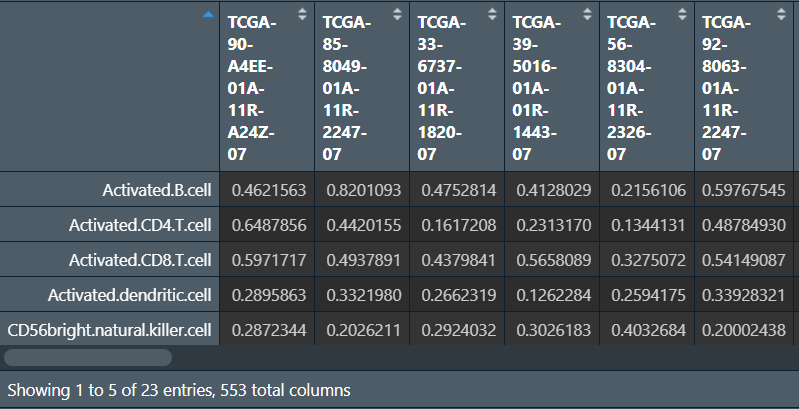
去除正常样本,只保留肿瘤样本,并与风险得分合并:
# 只保留肿瘤样本
group <- sapply(strsplit(colnames(ssgseaScore),"\\-"), "[", 4);
group <- sapply(strsplit(group,""), "[", 1);
group <- gsub("2", "1", group);
ssgseaScore2 <- t(ssgseaScore[, group==0]);
rownames(ssgseaScore2) <- gsub("(.*?)\\-(.*?)\\-(.*?)\\-(.*?)\\-.*", "\\1\\-\\2\\-\\3", rownames(ssgseaScore2));
# 合并
same_sample <- intersect(row.names(risk), row.names(ssgseaScore2));
risk <- risk[same_sample, "risk", drop=F]; # drop=F防止单列转为数组
rt2 <- ssgseaScore2[same_sample, ,drop=F];
rt1 <- cbind(rt2, risk);
rt <- melt(rt1, id.vars = c("risk")); # 宽数据->长数据
colnames(rt) <- c("Risk", "Type", "Score");
rt1:
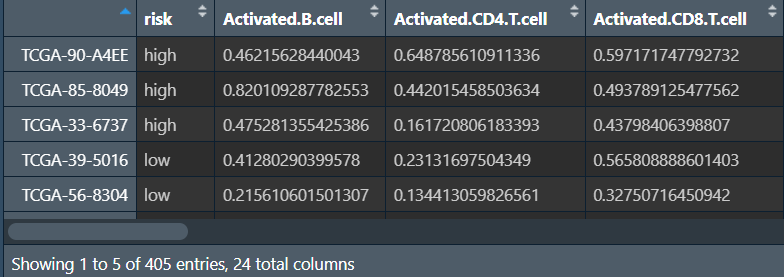
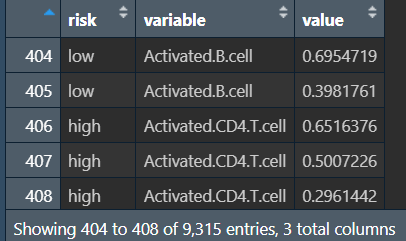
rt:将rt1的risk风险值列保留(作为分组依据),其余各免疫细胞名列都转为免疫细胞名-对应得分的两列
画图(高/低风险组的各免疫细胞得分差别):
rt$Risk <- factor(rt$Risk, levels=c("low","high")); # 按高低风险分组
p <- ggboxplot(
rt, x = "Type", y = "Score",
color = "Risk",
ylab = "Score",
add = "none",
xlab = "",
palette = c("Orange2", "DarkOrchid")
) +
rotate_x_text(50) +
stat_compare_means(
aes(group = Risk),
symnum.args = list(
cutpoints = c(0, 0.001, 0.01, 0.05, 1),
symbols = c("***", "**", "*", "")
),
label = "p.signif"
);
pdf(file="save_data\\immune_ssGSEA.pdf", width = 8, height = 5);
print(p);
dev.off();
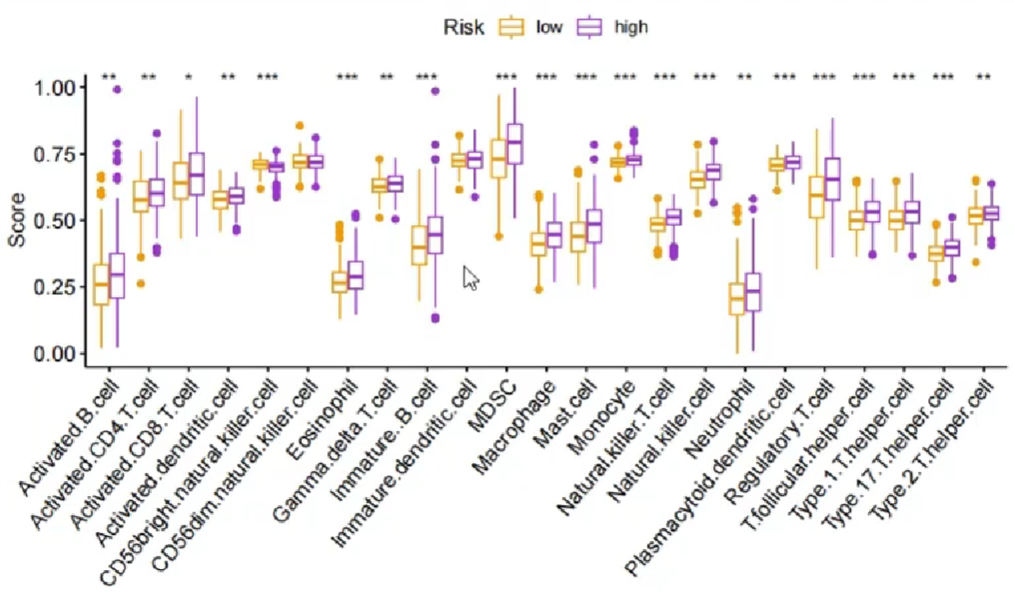
纵坐标是免疫细胞的相对得分,横坐标是免疫细胞种类,分组是按高/低风险,也可改成基因的表达高低
免疫功能分析(ssGSEA)
过程与上面的基本相同,只是使用的数据集不同,这里是免疫功能和其对应的基因:
-
读取tpm表达矩阵、数据集、风险得分
-
ssgsea分析
-
对ssGSEA score进行标注啊
-
去除正常样品
-
将风险得分与ssgsea分析结果合并
-
画图
library(limma);
library(GSVA);
library(GSEABase);
library(ggpubr);
library(reshape2);
# tpm表达矩阵
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", check.names = F, row.names = 1, sep = '\t', header = T);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
# 数据集文件
geneSet <- getGmt("data\\免疫数据\\immune2.gmt", geneIdType = SymbolIdentifier());
# 风险得分
risk <- read.table( "save_data\\risk.txt", check.names = F, row.names = 1, sep = '\t', header = T);
# ssGSEA分析
gsvapar <- gsvaParam(data, geneSet, kcdf = 'Gaussian', absRanking = TRUE);
ssgseaScore <- gsva(gsvapar);
normalize <- function(x){
return((x-min(x))/(max(x)-min(x)));
}
# 对ssGSEA score进行矫正
ssgseaScore <- normalize(ssgseaScore);
ssgseaOut <- rbind(id = colnames(ssgseaScore), ssgseaScore);
write.table(ssgseaOut, file="save_data\\immScore2.txt", sep="\t", quote=F, col.names=F);
# 只保留肿瘤样本
group <- sapply(strsplit(colnames(ssgseaScore),"\\-"), "[", 4);
group <- sapply(strsplit(group,""), "[", 1);
group <- gsub("2", "1", group);
ssgseaScore2 <- t(ssgseaScore[, group==0]);
rownames(ssgseaScore2) <- gsub("(.*?)\\-(.*?)\\-(.*?)\\-(.*?)\\-.*", "\\1\\-\\2\\-\\3", rownames(ssgseaScore2));
# 合并
same_sample <- intersect(row.names(risk), row.names(ssgseaScore2));
risk <- risk[same_sample, "risk", drop=F];
rt2 <- ssgseaScore2[same_sample, ,drop=F];
rt1 <- cbind(rt2, risk);
rt <- melt(rt1, id.vars = c("risk"));
colnames(rt) <- c("Risk", "Type", "Score");
# 画图
rt$Risk <- factor(rt$Risk, levels=c("low","high")); # 按高低风险分组
p <- ggboxplot(
rt, x = "Type", y = "Score",
color = "Risk",
ylab = "Score",
add = "none",
xlab = "",
palette = c("Orange2", "DarkOrchid")
) +
rotate_x_text(50) + # 旋转横坐标
stat_compare_means( # 将均值比较后的P值添加到ggplot图形中
aes(group = Risk),
symnum.args = list(
cutpoints = c(0, 0.001, 0.01, 0.05, 1),
symbols = c("***", "**", "*", "")
),
label = "p.signif"
);
pdf(file="save_data\\immune_ssGSEA2.pdf", width = 8, height = 5);
print(p);
dev.off();
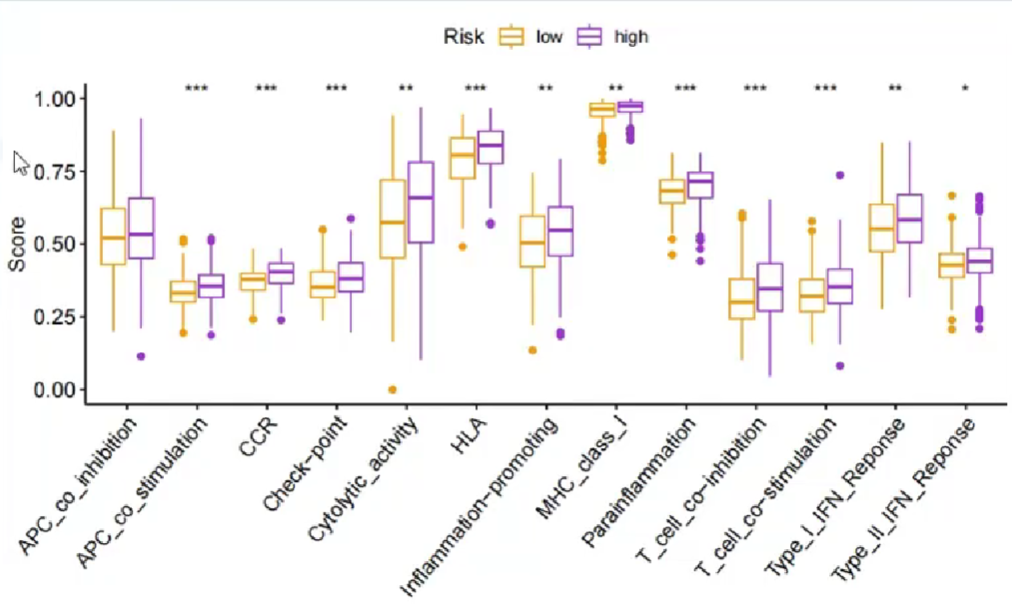
与上面的免疫细胞浸润分析类似,只不过横坐标是免疫功能