b站生信课程02-4
r-bioinfolessonb站生信课程TCGA及GEO数据挖掘入门必看P27-P34笔记——突变数据处理、预测免疫治疗反应和药物敏感性、免疫浸润评分、临床信息相关性分析、GEO差异表达分析
写在前面:本篇教程来自b站课程TCGA及GEO数据挖掘入门必看 P27-P34 相关资料下载
突变数据整理
if(!require("maftools", quietly = T))
{
library("BiocManager");
BiocManager::install("maftools");
library("maftools");
}
library(dplyr);
合并所有数据文件:
wd <- "data/突变数据/gdc_download_20231124_162149.342756/";
files <- list.files(wd, pattern = '*.gz', recursive = TRUE);
all_mut <- data.frame();
for (file in files) {
file <- paste0(wd, file);
mut <- read.delim(file, skip = 7, header = T, fill = TRUE, sep = "\t");
all_mut <- rbind(all_mut,mut);
}
# 样本名仅保留前12个字符
all_mut$Tumor_Sample_Barcode <- substr(all_mut$Tumor_Sample_Barcode, 1, 12);
# 数据读入
all_mut <- read.maf(all_mut);
# 选取指定列,更改样本名
a <- all_mut@data %>%
.[,c("Hugo_Symbol","Variant_Classification","Tumor_Sample_Barcode")] %>%
as.data.frame() %>%
mutate(Tumor_Sample_Barcode = substring(.$Tumor_Sample_Barcode,1,12));
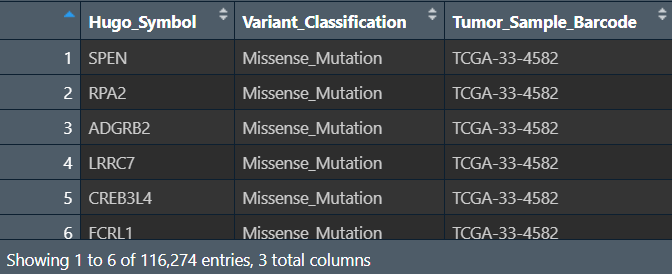
每个样本的每个基因都发生了什么样的突变
创建两个矩阵:列名是样本名,行名是基因名,值分别是该样本的该基因是否发生突变、发生了什么样的突变(如果未发生突变就为NA)
gene <- as.character(unique(a$Hugo_Symbol)); # 所有基因名
sample <- as.character(unique(a$Tumor_Sample_Barcode)); # 所有样本名
# 发生了什么样的突变
mat <- as.data.frame( # 列名是样本名,行名是基因名的空矩阵
matrix(
"",
length(gene),length(sample),
dimnames = list(gene,sample)
)
);
for (i in 1:nrow(a)){
mat[as.character(a[i,1]),as.character(a[i,3])] <- as.character(a[i,2]); # 根据行列名设定值
}
# 是否发生突变
mat_0_1 <- as.data.frame( # 列名是样本名,行名是基因名的空矩阵
matrix(
0,
length(gene),length(sample),
dimnames = list(gene,sample)
)
);
for (i in 1:nrow(a)){
mat_0_1[as.character(a[i,1]),as.character(a[i,3])] <- 1; # 没取到的位置就为NA
}
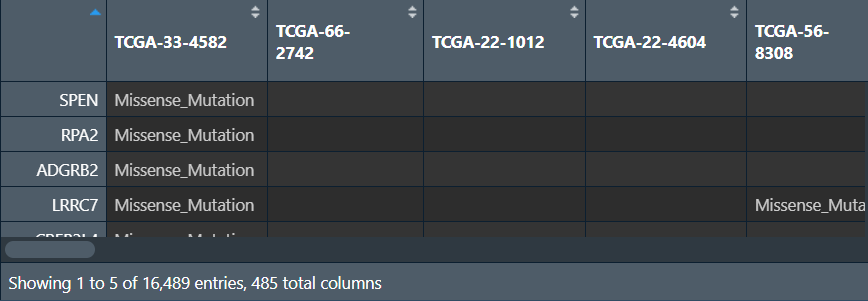
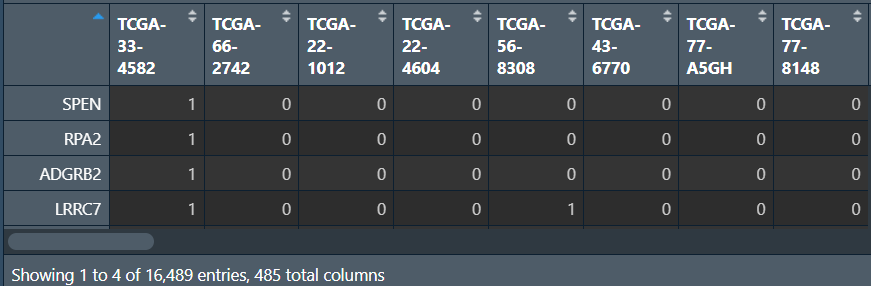
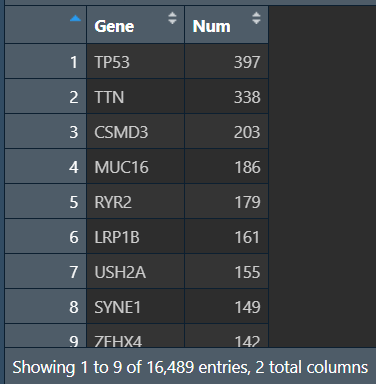
汇总所有样本的突变情况(每个基因在多少个样本中发生了突变):将行名变成Gene列,对应的count列只需对mat_0_1的每行加和即可(有突变就是1,有几个1就有几个突变)
gene_count <- data.frame(
Gene = rownames(mat_0_1),
Num = as.numeric(apply(mat_0_1, 1, sum))
) %>%
arrange(desc(Num)); # 按突变数从大到小排序
# 保存数据
write.table(gene_count, 'save_data\\geneMut.txt', sep="\t", quote=F, row.names = F);
write.mafSummary(maf = all_mut, basename = "save_data\\input");
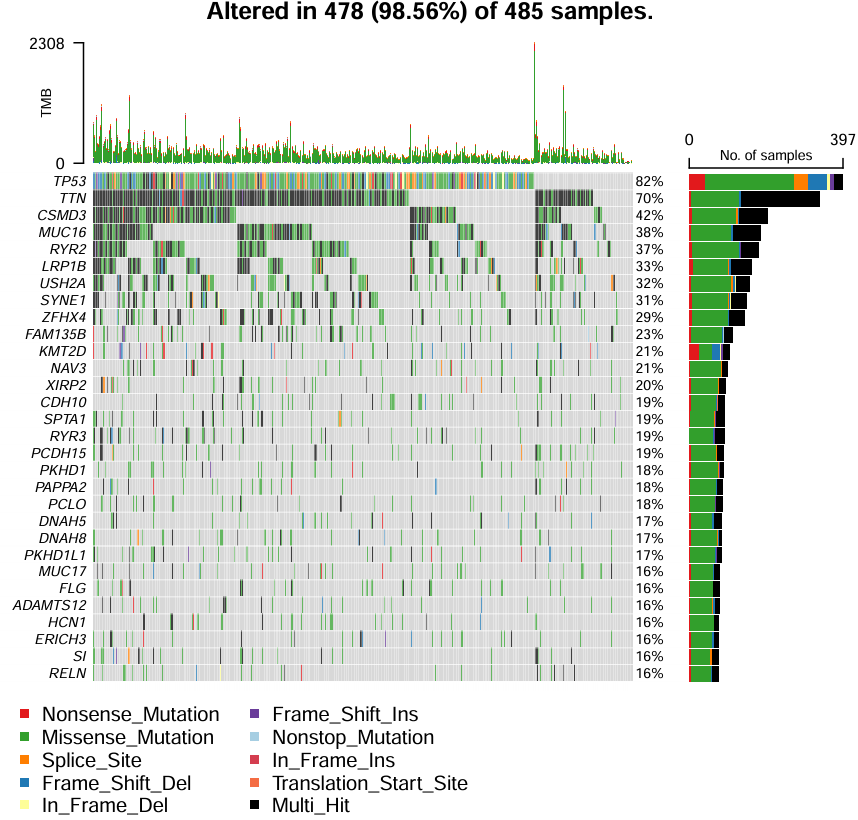
绘制瀑布图oncoplot:
pdf(file = "save_data\\maf.pdf", width = 6, height = 6);
oncoplot(
maf = all_mut,
top = 30, # 显示前30个的突变基因信息
fontSize = 0.6, # 设置字体大小
showTumorSampleBarcodes = F
); # 不显示病人信息
dev.off();
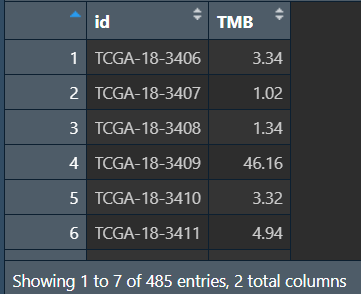
计算每个样本的TMB值(肿瘤突变负荷:每百万碱基检测出的体细胞变异总数)
tmb_table <- tmb(maf = all_mut, logScale = F);
tmb_table <- tmb_table[, c(1,3)]; # 保留需要的tmb值信息
tmb_table <- as.data.frame(tmb_table);
tmb_table[,1] <- substr(tmb_table[,1],1,12);
tmb_table <- aggregate( . ~ Tumor_Sample_Barcode, data = tmb_table, max); # 去重,重复行取最大值
colnames(tmb_table)[1] = "id";
colnames(tmb_table)[2] = "TMB";
write.table(tmb_table,'save_data\\TMB.txt', sep="\t", quote=F, row.names = F);
TIDE预测免疫治疗反应
TIDE:肿瘤免疫功能障碍和排斥,用于评估肿瘤样本基因表达谱中肿瘤免疫逃逸的可能性——值越大,越可能免疫,对免疫治疗越不敏感
library(limma);
library(ggpubr);
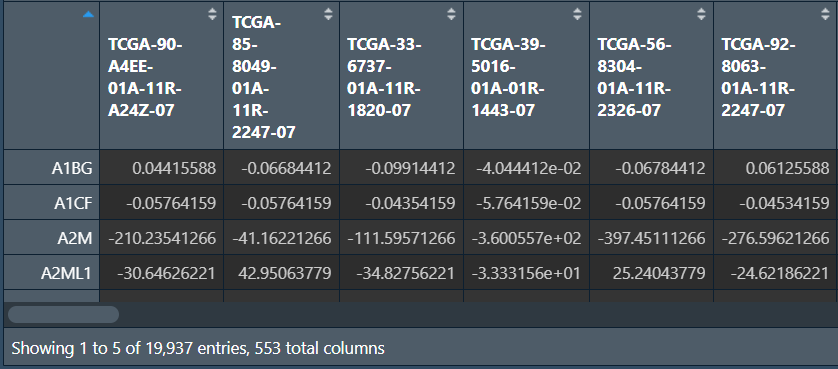
读取tpm表达矩阵,并进行标准化:数据值-行平均值,使基因的均值为0
rt <- read.table("save_data\\TCGA_LUSC_TPM.txt", header = T, sep = "\t", check.names = F);
rownames(rt) <- rt[, 1];
exp <- rt[, 2:ncol(rt)];
Expr <- t(apply(exp, 1, function(x)x-(mean(x))));
write.table(
data.frame(ID = rownames(Expr), Expr, check.names = F),
'save_data\\tcga_normalize.txt', sep="\t", quote=F, row.names = TRUE
);
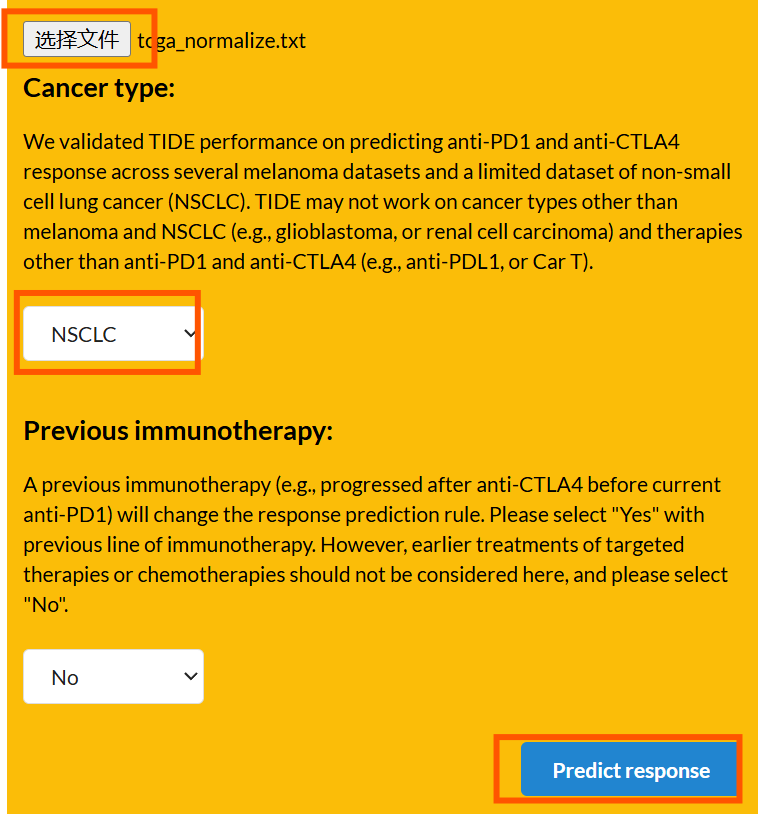
TIDE数据的获取:进入网站
-
点击
选择文件,将刚才得到的标准化表达矩阵放入 -
cancer type:NSCLC -
点击
Predict response
之后点击页面最下面的Export to CSV导出
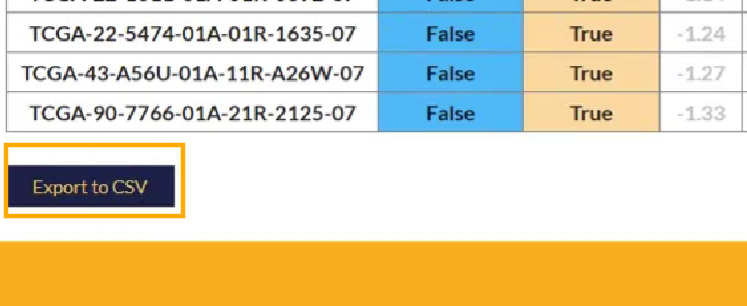
只需要两列信息:patient样本名、TIDE
读取TIDE数据并处理:
tide <- read.csv("save_data\\TIDE.csv");
tide <- tide[, c(1, 4)];
# 仅保留肿瘤样本
group <- sapply(strsplit(tide[, 1],"\\-"), "[", 4);
group <- sapply(strsplit(group,""), "[", 1);
group <- gsub("2", "1", group);
tide <- tide[group==0, , drop=F];
# 修改样本名
tide[, 1] <- gsub("(.*?)\\-(.*?)\\-(.*?)\\-(.*?)\\-.*", "\\1\\-\\2\\-\\3", tide[, 1]);
# 去除重复
tide1 <- tide[!duplicated(tide$Patient), ];
# 让样本名为行名
rownames(tide1) <- tide1[, 1];
tide <- as.data.frame(tide1[, 2]);
rownames(tide) <- rownames(tide1);
colnames(tide) <- "TIDE";
tide <- avereps(tide);
读取风险得分,根据样本名合并,同时设置比较组:
-
更改risk列:如果值为high就改为”High-risk”,反之为”Low-risk”
-
将所有的组名两两组合,存入一个列表中,使每个元素是一个有两个元素(组名)的字符串数组
# 读取风险得分
risk <- read.table( "save_data\\risk.txt", check.names = F, row.names = 1, sep = '\t', header = T);
# 合并
same_sample <- intersect(row.names(risk), row.names(tide));
data <- cbind(
tide[same_sample, , drop=F],
risk[same_sample, "risk", drop=F]
);
# 设置比较组
data$risk <- ifelse(data$risk=="high", "High-risk", "Low-risk");
group <- levels(factor(data$risk));
comp <- combn(group, 2); # 将group中每2个元素进行组合
my_comparisons <- list();
for(i in 1:ncol(comp)){
my_comparisons[[i]] <- comp[, i];
}
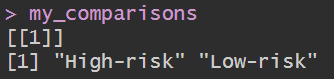
画图:
pdf(file = "save_data\\TIDE.pdf", width = 5, height = 4.5);
ggviolin(
data,
x = "risk", y="TIDE",
fill = "risk",
xlab = "", ylab = "TIDE",
palette = c("Firebrick2", "DodgerBlue1"),
legend.title = "Risk",
add = "boxplot",
add.params = list(fill = "white")
) +
stat_compare_means(comparisons = my_comparisons); # 添加p值显著性标记
dev.off();
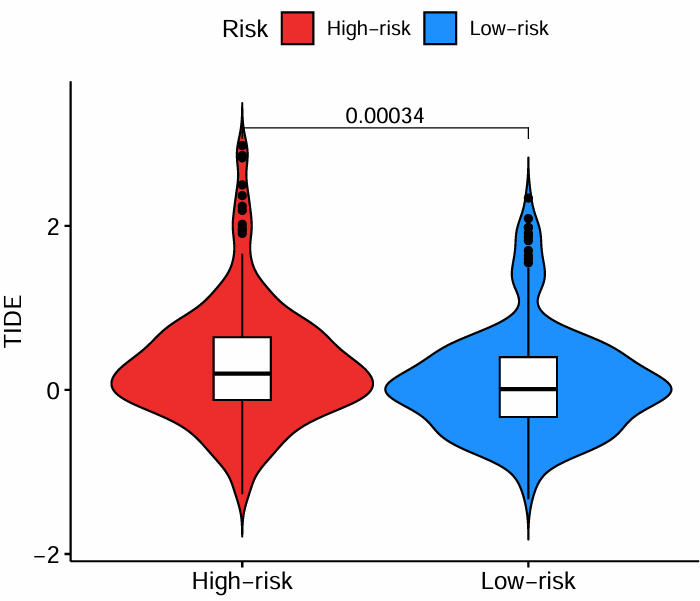
横坐标是高低风险组,纵坐标是TIDE得分。可以看到低风险组的TIDE低于高风险组,对免疫治疗更敏感
预测药物敏感性
oncoPredict
药物预测需要训练集,一般推荐使用权威数据作为训练集建模,之后预测自己的数据。这里使用GDSC(Genomics of Drug Sensitivity in Cancer)数据库
使用oncoPredict包
需要数据:tpm表达矩阵、风险得分、2个GDSC文件
if(!require("GenomicFeatures", quietly = T))
{
library("BiocManager");
BiocManager::install("GenomicFeatures");
library("GenomicFeatures");
}
if(!require("TCGAbiolinks", quietly = T))
{
library("BiocManager");
BiocManager::install("TCGAbiolinks");
library("TCGAbiolinks");
}
if(!require("TxDb.Hsapiens.UCSC.hg19.knownGene", quietly = T))
{
library("BiocManager");
BiocManager::install("TxDb.Hsapiens.UCSC.hg19.knownGene");
library("TxDb.Hsapiens.UCSC.hg19.knownGene");
}
if(!require("oncoPredict", quietly = T))
{
install.packages("oncoPredict");
}
library(limma);
library(oncoPredict);
library(parallel);
library(limma);
library(ggplot2);
library(ggpubr);
library(limma);
library(reshape2);
library(ggplot2);
library(ggpubr);
读取数据:
# tpm表达矩阵
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", header = T, sep = "\t", check.names = F, row.names = 1);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
colnames(data) <- gsub("(.*?)\\_(.*?)", "\\2", colnames(data));
# 读取风险得分
risk <- read.table( "save_data\\risk.txt", check.names = F, row.names = 1, sep = '\t', header = T);
读取GDSC文件并建模:
set.seed(999); # 随机数种子
# 读取GDSC文件
GDSC2_Expr <- readRDS(file = 'data\\免疫数据\\GDSC2_Expr.rds');
GDSC2_Res <- readRDS(file = 'data\\免疫数据\\GDSC2_Res.rds');
GDSC2_Res <- exp(GDSC2_Res);
# 药物敏感性建模(半小时左右)
calcPhenotype(
trainingExprData = GDSC2_Expr,
trainingPtype = GDSC2_Res,
testExprData = data,
batchCorrect = 'eb',
powerTransformPhenotype = TRUE,
removeLowVaryingGenes = 0.2,
minNumSamples = 10,
printOutput = TRUE,
removeLowVaringGenesFrom = 'rawData'
);
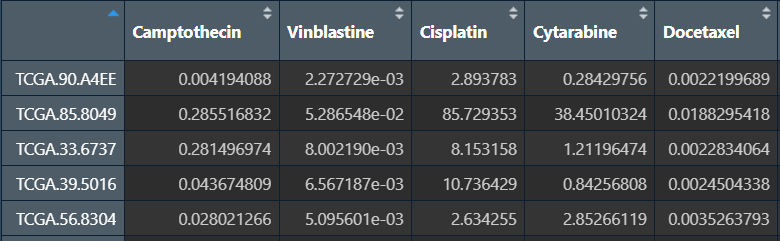
横坐标是样本,纵坐标是药物名,显示不同药物在每一个样本中的敏感性
读入药敏文件,处理后与风险得分合并:
# 读入药敏文件
senstivity <- read.csv("save_data\\calcPhenotype_Output\\DrugPredictions.csv", header = T, sep = ",", check.names = F, row.names = 1);
colnames(senstivity) <- gsub("(.*)\\_(\\d+)", "\\1", colnames(senstivity));
# 仅保留肿瘤样本
group <- sapply(strsplit(rownames(senstivity),"\\-"), "[", 4);
group <- sapply(strsplit(group,""), "[", 1);
senstivity <- senstivity[group==0,];
# 样本名仅保留前12字符
senstivity <- t(senstivity); # 为避免行名重复,先转置,改变列名
colnames(senstivity) <- substr(colnames(senstivity), 1, 12);
senstivity <- t(senstivity); # 最后再转回来
# 合并
same_sample <- intersect(row.names(risk), row.names(senstivity));
senstivity <- senstivity[same_sample, , drop=F];
risk <- risk[same_sample, "risk", drop=F];
# NA->0
senstivity[is.na(senstivity)] <- 0;
# 取log2
senstivity <- log2(senstivity+1);
rt <- cbind(risk, senstivity);
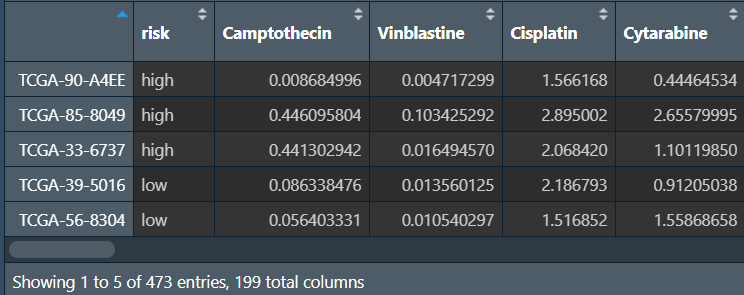
设置比较组、提取显著差异的药物:
# 设置比较组
rt$risk <- factor(rt$risk, levels = c("low", "high"));
group <- levels(factor(rt$risk));
comp <- combn(group, 2);
my_comparisons <- list();
for(i in 1:ncol(comp)){
my_comparisons[[i]] <- comp[, i];
}
# 提取显著差异的药物
sigGene <- c();
for(i in colnames(rt)[2:(ncol(rt))]){
if(sd(rt[, i])<0.05){next} # 某列(某种药物)的标准差<0.05就跳过
wilcoxTest <- wilcox.test(rt[, i] ~ rt[, "risk"]);
pvalue <- wilcoxTest$p.value;
if(wilcoxTest$p.value<0.001){ # 如果药物数量较少,可设为0.05
sigGene <- c(sigGene, i);
}
}
sigGene <- c(sigGene, "risk");
rt_sig <- rt[, sigGene];
绘图前的数据准备:
# 宽数据变长数据(risk列不变,其它列名变列值
rt_sig <- melt(rt_sig,id.vars=c("risk"));
colnames(rt_sig) <- c("risk","Gene","Expression");
# 设置比较组
group <- levels(factor(rt$risk));
rt_sig$risk <- factor(rt_sig$risk, levels = c("low", "high"));
comp <- combn(group, 2);
my_comparisons <- list();
for(i in 1:ncol(comp)){
my_comparisons[[i]] <- comp[, i];
}
画箱型图:
boxplot <- ggboxplot(
rt_sig, x = "Gene", y = "Expression",
fill = "risk",
xlab = "", ylab = "Drug Senstivity",
legend.title = "Risk",
width = 0.8,
palette = c("DodgerBlue1","Firebrick2")
) +
rotate_x_text(50) +
stat_compare_means(
aes(group = risk),
method = "wilcox.test",
symnum.args = list(
cutpoints = c(0, 0.001, 0.01, 0.05, 1),
symbols = c("***", "**", "*", "ns")
),
label = "p.signif"
) +
theme(
axis.text = element_text(
face = "bold.italic",
colour = "#441718",
size = 16
),
axis.title = element_text(
face = "bold.italic",
colour = "#441718",
size = 16
),
axis.line = element_blank(),
plot.title = element_text(
face = "bold.italic",
colour = "#441718",
size = 16
),
legend.text = element_text(face = "bold.italic"),
panel.border = element_rect(
fill = NA,
color = "#35A79D",
size = 1.5,
linetype = "solid"
),
panel.background = element_rect(fill = "#F1F6FC"),
panel.grid.major = element_line(
color = "#CFD3D6",
size = .5,
linetype = "dotdash"
),
legend.title = element_text(
face = "bold.italic",
size = 13
)
);
# 输出图片
pdf(file = "save_data\\drugSenstivity.pdf", width = 20, height = 8);
print(boxplot);
dev.off();
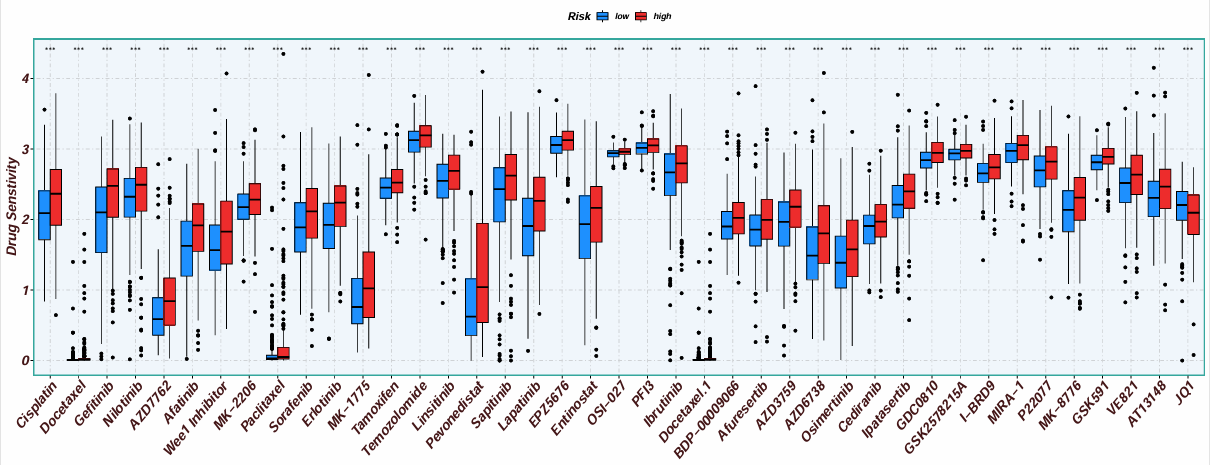
横坐标是不同的药物,纵坐标是药物敏感性,按高/低风险分组(也可以是某个基因的表达量高低)。得分越低,则该药物在该组中越敏感
pRRophetic
使用pRRophetic包,它比上面的oncoPredict包旧一点,所以要使用R<4.3版本运行,推荐使用上面的oncoPredict包
install.packages("C:\\Users\\WangTianHao\\Downloads\\pRRophetic_0.5.tar.gz", repos = NULL, type = "source");
library(limma);
library(ggpubr);
library(pRRophetic);
library(ggplot2);
set.seed(12345);
读入数据:tpm表达矩阵和风险分组
# 表达矩阵
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", header = T, sep = "\t", check.names = F,row.names = 1);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
data <- data[rowMeans(data)>0.5, ];
#仅保留肿瘤样本
group <- sapply(strsplit(colnames(data),"\\-"), "[", 4);
group <- sapply(strsplit(group, ""), "[", 1);
group <- gsub("2", "1", group);
data <- data[, group==0];
data <- t(data);
rownames(data) <- gsub("(.*?)\\-(.*?)\\-(.*?)\\-(.*)", "\\1\\-\\2\\-\\3", rownames(data));
data <- avereps(data);
data <- t(data);
# 风险分组
riskRT <- read.table("save_data\\risk.txt", header = T, sep = "\t", check.names = F, row.names = 1);
使用CGP数据库,使用?pRRopheticPredict–arguments–drug可以看到它可以预测哪些药物
进行预测:
# 能预测的药物
alldrugs <- c("A.443654","A.770041","ABT.263","ABT.888","AG.014699","AICAR","AKT.inhibitor.VIII","AMG.706","AP.24534","AS601245","ATRA","AUY922","Axitinib","AZ628","AZD.0530","AZD.2281","AZD6244","AZD6482","AZD7762","AZD8055","BAY.61.3606","Bexarotene","BI.2536","BIBW2992","Bicalutamide","BI.D1870","BIRB.0796","Bleomycin","BMS.509744","BMS.536924","BMS.708163","BMS.754807","Bortezomib","Bosutinib","Bryostatin.1","BX.795","Camptothecin","CCT007093","CCT018159","CEP.701","CGP.082996","CGP.60474","CHIR.99021","CI.1040","Cisplatin","CMK","Cyclopamine","Cytarabine","Dasatinib","DMOG","Docetaxel","Doxorubicin","EHT.1864","Elesclomol","Embelin","Epothilone.B","Erlotinib","Etoposide","FH535","FTI.277","GDC.0449","GDC0941","Gefitinib","Gemcitabine","GNF.2","GSK269962A","GSK.650394","GW.441756","GW843682X","Imatinib","IPA.3","JNJ.26854165","JNK.9L","JNK.Inhibitor.VIII","JW.7.52.1","KIN001.135","KU.55933","Lapatinib","Lenalidomide","LFM.A13","Metformin","Methotrexate","MG.132","Midostaurin","Mitomycin.C","MK.2206","MS.275","Nilotinib","NSC.87877","NU.7441","Nutlin.3a","NVP.BEZ235","NVP.TAE684","Obatoclax.Mesylate","OSI.906","PAC.1","Paclitaxel","Parthenolide","Pazopanib","PD.0325901","PD.0332991","PD.173074","PF.02341066","PF.4708671","PF.562271","PHA.665752","PLX4720","Pyrimethamine","QS11","Rapamycin","RDEA119","RO.3306","Roscovitine","Salubrinal","SB.216763","SB590885","Shikonin","SL.0101.1","Sorafenib","S.Trityl.L.cysteine","Sunitinib","Temsirolimus","Thapsigargin","Tipifarnib","TW.37","Vinblastine","Vinorelbine","Vorinostat","VX.680","VX.702","WH.4.023","WO2009093972","WZ.1.84","X17.AAG","X681640","XMD8.85","Z.LLNle.CHO","ZM.447439");
# 进行预测并画图(可能需要半小时左右)
for(drug in alldrugs){
# 预测药物敏感性
sensitivity <- pRRopheticPredict(data, drug, selection = 1);
sensitivity <- sensitivity[sensitivity!="NaN"];
# 合并风险文件和药物敏感性结果
sameSample <- intersect(row.names(riskRT), names(sensitivity));
risk <- riskRT[sameSample, "risk", drop=F];
sensitivity <- sensitivity[sameSample];
rt <- cbind(risk, sensitivity);
# 设置比较组
rt$risk <- factor(rt$risk, levels = c("low", "high"));
type <- levels(factor(rt[, "risk"]));
comp <- combn(type, 2);
my_comparisons <- list();
for(i in 1:ncol(comp)){
my_comparisons[[i]]<-comp[,i];
}
#获取差异pvalue
test <- wilcox.test(sensitivity ~ risk, data = rt);
if(test$p.value < 0.05){ # 如果想得到更少的结果,就改成0.001
#绘制箱线图
boxplot <- ggboxplot(
rt, x = "risk", y = "sensitivity",
fill = "risk",
xlab = "Risk", ylab = paste0(drug, " sensitivity (IC50)"),
legend.title = "Risk",
palette = c("DodgerBlue1", "Firebrick2")
) +
stat_compare_means(comparisons = my_comparisons);
pdf(file = paste0("save_data\\pRRopheticPredict\\durgsensitivity.", drug, ".pdf"), width = 2.3, height = 4.3);
print(boxplot);
dev.off();
}
}
注:这种方法是每个药物都画一张图
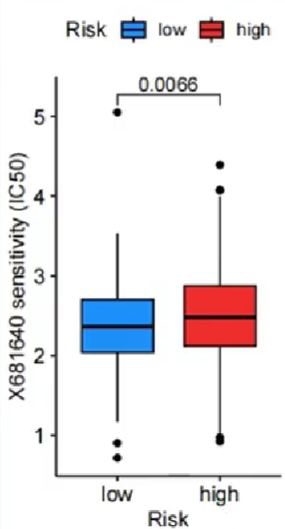
横坐标是不同的分组(高/低风险),纵坐标是药物敏感性。与上面方法相同,药物敏感性值越低,对这个药物越敏感
免疫浸润评分(estimate)
if(!require("estimate", quietly = T))
{
library(utils);
rforge <- "http://r-forge.r-project.org";
install.packages("estimate", repos = rforge, dependencies = TRUE);
}
library(limma);
library(estimate);
library(reshape2);
library(ggpubr);
进行分析:
-
基因过滤,将表达矩阵中的基因和
estimate包的common gene(存储在commonGenes.gct中)进行比对过滤 -
运行
estimate包
# 基因过滤
filterCommonGenes(
input.f = "save_data\\TCGA_LUSC_TPM.txt",
output.f = "data\\免疫数据\\commonGenes.gct",
id = "GeneSymbol"
);
# 运行estimate包
estimateScore(
input.ds = "data\\免疫数据\\commonGenes.gct",
output.ds = "save_data\\estimateScore.gct" # 输出结果
);
# 保存每个样品的打分
scores <- read.table("save_data\\estimateScore.gct", skip = 2, header = T);
rownames(scores) <- scores[, 1];
scores <- t(scores[, 3:ncol(scores)]);
rownames(scores) <- gsub("\\.", "\\-", rownames(scores));
out <- rbind(ID = colnames(scores), scores);
write.table(out, file = "save_data\\TMEscores.txt", sep = "\t", quote = F, col.names = F);
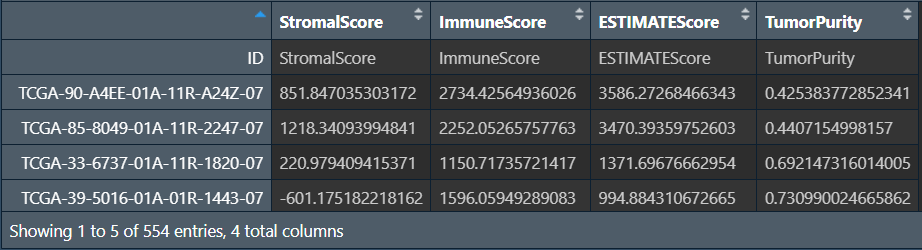
这四列分别是StromalScore肿瘤组织中的基质细胞、ImmuneScore肿瘤组织中的免疫细胞浸润、ESTIMATEScoreestimate得分、TumorPurity肿瘤纯度
其中ESTIMATEScore = StromalScore + ImmuneScore,该值与肿瘤纯度成负相关(ESTIMATEScore越高,肿瘤纯度越低)
ESTIMATEScore越高,肿瘤微环境中基质细胞和免疫细胞含量越高,肿瘤细胞含量(肿瘤纯度)越低,预后越好
画图前的数据处理:
-
仅保留estimate结果的前3列(
TumorPurity的值与另三个值差距太大,一起画图不好统一坐标轴,且它与Estimate socre成负相关,可单独画图) -
读取风险分组,并获取共同样本,合并
-
宽数据->长数据(同前)
# 仅保留前3列
score <- scores[,1:3];
# 读取风险文件
Risk <- read.table("save_data\\risk.txt", header = T, sep = "\t", check.names = F, row.names = 1);
Risk$risk <- factor(Risk$risk, levels = c("low","high"));
# 仅保留肿瘤样本
group <- sapply(strsplit(rownames(score), "\\-"), "[", 4);
group <- sapply(strsplit(group, ""), "[", 1);
score <- score[group==0, ];
rownames(score) <- substr(rownames(score), 1, 12);
rownames(score) <- gsub('[.]', '-', rownames(score));
# 获取共同样本
score <- score[row.names(Risk), , drop=F];
# 合并
rt <- cbind(Risk[,"risk", drop=F], score);
# 宽变长
data <- melt(rt, id.vars = c("risk"));
colnames(data) <- c("Risk", "scoreType", "Score");
画小提琴图:
p <- ggviolin(
data, x = "scoreType", y = "Score",
fill = "Risk",
xlab = "", ylab = "TME score",
legend.title = "Risk",
add = "boxplot",
add.params = list(color = "white"),
palette = c("SteelBlue3", "Firebrick3", "green"),
width = 1
);
p <- p + rotate_x_text(45);
p1 <- p + stat_compare_means(
aes(group = Risk),
method = "wilcox.test",
symnum.args = list(
cutpoints = c(0, 0.001, 0.01, 0.05, 1),
symbols = c("***", "**", "*", " ")
),
label = "p.signif"
);
#输出图形
pdf(file = "save_data\\TME.vioplot.pdf", width = 8, height = 5);
print(p1);
dev.off();
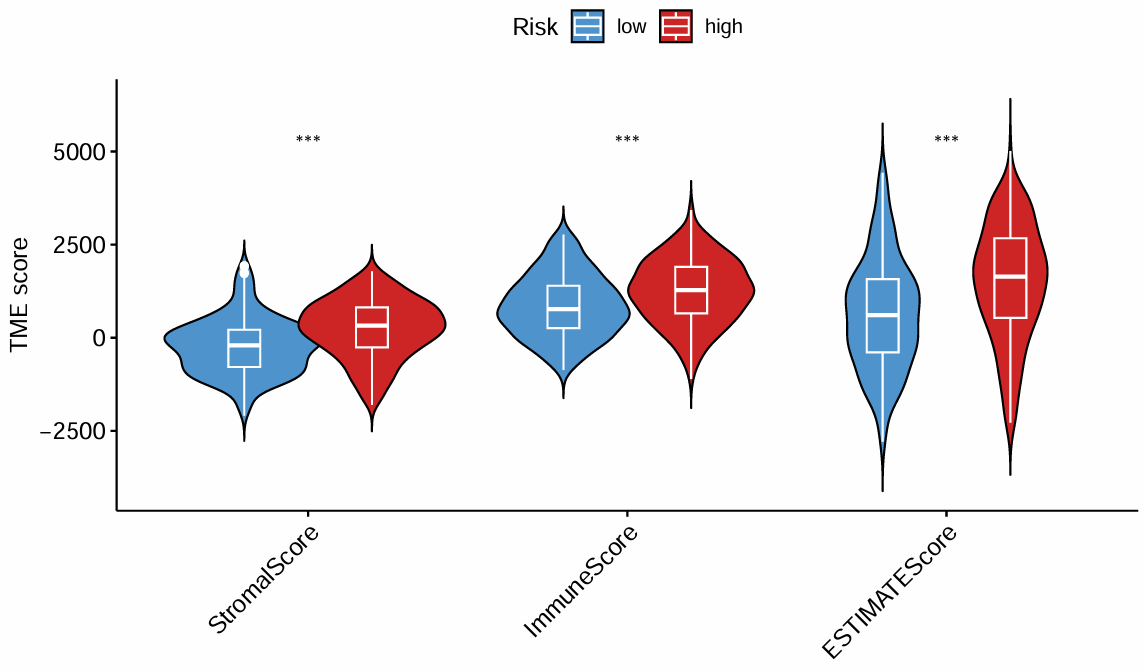
按高/低风险分组,横纵坐标是三种得分
临床信息相关性分析
热图
if(!require("ComplexHeatmap", quietly = T))
{
library("BiocManager");
BiocManager::install("ComplexHeatmap");
library("ComplexHeatmap");
}
library(limma);
读取数据:tpm表达矩阵、临床信息
-
去除正常样品
-
根据某个基因表达量的中位值(这里以ABCA3为例),把样品分为两组
-
把临床信息中的连续变量(如年龄等)通过分段的方式改成离散变量
-
合并
# tpm表达矩阵
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", header = T, sep = "\t", check.names = F, row.names = 1);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
# 仅保留肿瘤样本
group <- sapply(strsplit(colnames(data),"\\-"), "[", 4);
group <- sapply(strsplit(group, ""), "[", 1);
group <- gsub("2", "1", group);
data <- t(data[, group==0]);
rownames(data) <- gsub("(.*?)\\-(.*?)\\-(.*?)\\-(.*?)\\-.*", "\\1\\-\\2\\-\\3", rownames(data));
# 分组
gene <- "ABCA3";
Type <- ifelse(data[, gene]>quantile(data[, gene], seq(0, 1, 1/2))[2], "High", "Low");
Type <- factor(Type, levels = c("Low", "High"));
data <- cbind(as.data.frame(data), Type);
data <- data[, c(gene, "Type")];
rownames(data) <- gsub("[.]", "-", rownames(data));
data <- data[order(data[, gene]), ]; # 按表达量排序
# 临床信息
cli <- read.table("save_data\\clinical.txt", header = T, sep = "\t", check.names = F, row.names = 1);
cli[,"Age"] <- ifelse(
cli[, "Age"]=="unknow",
"unknow",
ifelse(cli[, "Age"]>65, ">65", "<=65")
);
# 合并
samSample <- intersect(row.names(data), row.names(cli));
data <- data[samSample, "Type", drop=F];
cli <- cli[samSample, , drop=F];
rt <- cbind(data, cli);
进行分析:临床性状在高低表达组之间是否具有差异
sigVec <- c(gene);
for(clinical in colnames(rt[, 2:ncol(rt)])){ # 遍历每个临床特征
data <- rt[c("Type", clinical)];
colnames(data) <- c("Type", "clinical");
data <- data[(data[, "clinical"]!="unknow"), ]; # 去除unknown值
tableStat <- table(data);
stat <- chisq.test(tableStat); # 检测方法
pvalue <- stat$p.value;
Sig <- ifelse( # 分成3档
pvalue<0.001,
"***",
ifelse(
pvalue<0.01,
"**",
ifelse(
pvalue<0.05,
"*",
""
)
)
);
sigVec <- c(sigVec, paste0(clinical, Sig)); # 向列名后添加显著性标识
}
colnames(rt) <- sigVec;
绘制热图:
# 设置颜色
bioCol <- c("#0066FF","#FF9900","#FF0000","#ed1299", "#0dbc21", "#246b93", "#cc8e12", "#d561dd", "#c93f00", "#ce2523", "#f7aa5d", "#9ed84e", "#39ba30", "#6ad157", "#373bbf", "#a1ce4c", "#ef3bb6", "#d66551", "#1a918f", "#7149af", "#ff66fc", "#2927c4", "#57e559" ,"#8e3af4" ,"#f9a270" ,"#22547f", "#db5e92", "#4aef7b", "#e86502", "#99db27", "#e07233", "#8249aa","#cebb10", "#03827f", "#931635", "#ff523f", "#edd05e", "#6f25e8", "#0dbc21", "#167275", "#280f7a", "#6373ed", "#5b910f" ,"#7b34c1" ,"#0cf29a" ,"#d80fc1", "#dd27ce", "#07a301", "#ddd53e", "#391c82", "#2baeb5","#925bea", "#09f9f5", "#63ff4f");
colorList <- list(); # 一个列表,存储临床特征及其对应的颜色
colorList[[gene]] <- c("Low" = "Orange2", "High" = "DarkOrchid"); # 高低表达组的颜色
j <- 0; # 标识使用到了第几个颜色
for(cli in colnames(rt[, 2:ncol(rt)])){
cliLength <- length(levels(factor(rt[, cli]))); # 某个临床特征的长度
cliCol <- bioCol[(j+1):(j+cliLength)]; # 给这个临床特征分配颜色
j <- j+cliLength;
names(cliCol) <- levels(factor(rt[, cli])); # 给颜色命名
cliCol["unknow"] <- "grey75"; # 未知值的颜色
colorList[[cli]] <- cliCol; # 添加到结果列表中
}
# 画图
zero_row_mat <- matrix(nrow = 0, ncol = nrow(rt)); # 每个样本都有一列
ha <- HeatmapAnnotation(df = rt, col = colorList);
Hm <- Heatmap(zero_row_mat, top_annotation = ha);
# 保存图片
pdf(file = "save_data\\cli_heatmap.pdf", width = 7, height = 5);
draw(Hm, merge_legend = TRUE, heatmap_legend_side = "bottom", annotation_legend_side = "bottom");
dev.off();
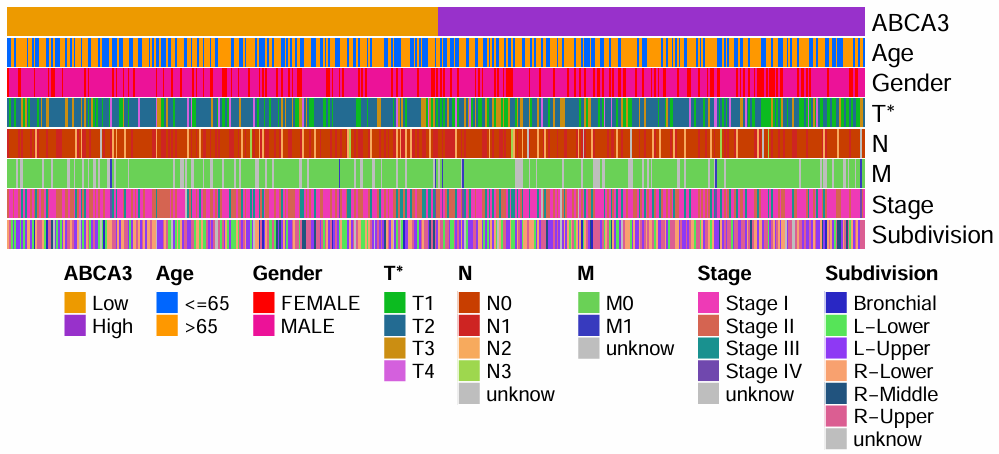
第一行是目标基因的表达量,根据这个表达量分为高/低表达组,下面是一些临床特征;特征名旁边的*代表该特征在高/低表达组中有显著差异,没有*就是没有显著差异
仔细观察,其实这个图纵向的每一个小竖线就代表一个样本
箱型图
读取数据:过程同上,只是没有分组的过程
library(limma);
library(ggpubr);
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", header = T, sep = "\t", check.names = F, row.names = 1);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
group <- sapply(strsplit(colnames(data),"\\-"), "[", 4);
group <- sapply(strsplit(group, ""), "[", 1);
group <- gsub("2", "1", group);
data <- t(data[, group==0]);
rownames(data) <- gsub("(.*?)\\-(.*?)\\-(.*?)\\-(.*?)\\-.*", "\\1\\-\\2\\-\\3", rownames(data));
cli <- read.table("save_data\\clinical.txt", header = T, sep = "\t", check.names = F, row.names = 1);
cli[,"Age"] <- ifelse(
cli[, "Age"]=="unknow",
"unknow",
ifelse(cli[, "Age"]>65, ">65", "<=65")
);
samSample <- intersect(row.names(data), row.names(cli));
data <- data[samSample, , drop=F];
cli <- cli[samSample, , drop=F];
rt <- cbind(data, cli);
rt <- rt[, c(gene, colnames(cli))];
画图:
for(clinical in colnames(rt[, 2:ncol(rt)])){
# 数据处理
data <- rt[c(gene, clinical)]; # 提取基因表达量和临床特征列
colnames(data) <- c(gene, "clinical"); # 更改列名
data <- data[(data[, "clinical"]!="unknow"), ]; # 去除未知值
group <- levels(factor(data$clinical));
data$clinical <- factor(data$clinical, levels = group); # 将临床特征列改为factor形式
comp <- combn(group,2); # 设置比较组,如果这里写成3,就是三组间相比
my_comparisons <- list();
for(i in 1:ncol(comp)){
my_comparisons[[i]]<-comp[,i];
}
# 绘制箱线图
boxplot <- ggboxplot(
data, x = "clinical", y = gene,
fill = "clinical",
xlab = clinical, ylab = paste(gene, " expression"),
legend.title = clinical
) +
stat_compare_means(comparisons = my_comparisons);
# 保存图片
pdf(file = paste0("save_data\\clinicalCor_", clinical, ".pdf"), width = 5.5, height = 5);
print(boxplot);
dev.off();
}
注:这种方法是每个临床特征画一张图
clinicalCor_T.pdf:
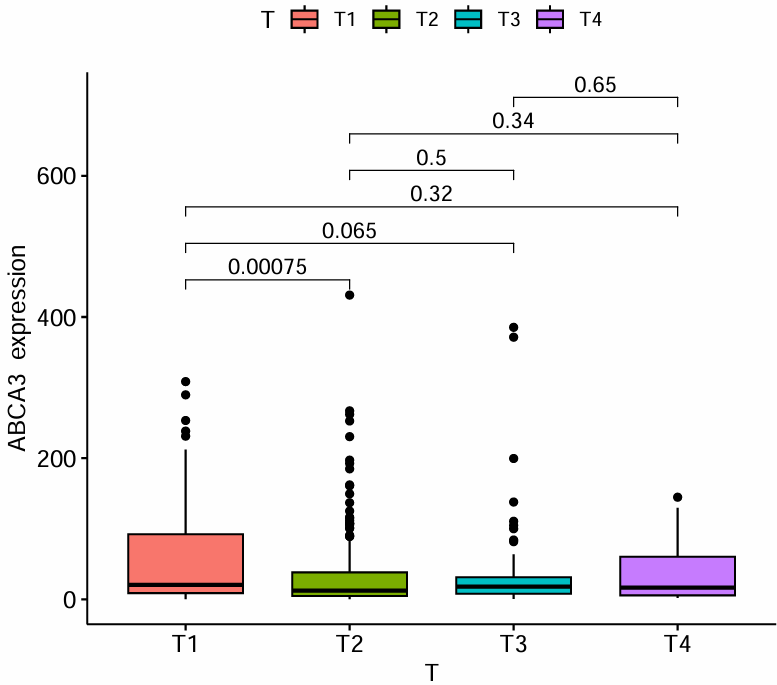
clinicalCor_Age.pdf:
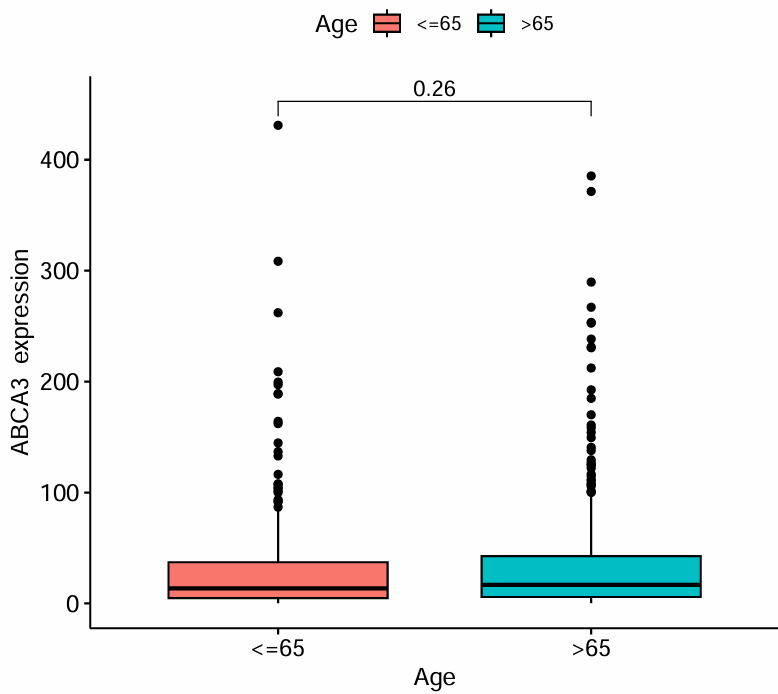
横坐标代表不同的T分期/年龄,纵坐标是目标基因的表达水平,上面的p值代表目标基因的表达水平在各分期/年龄间是否有统计学差异
差异表达分析(GEO)
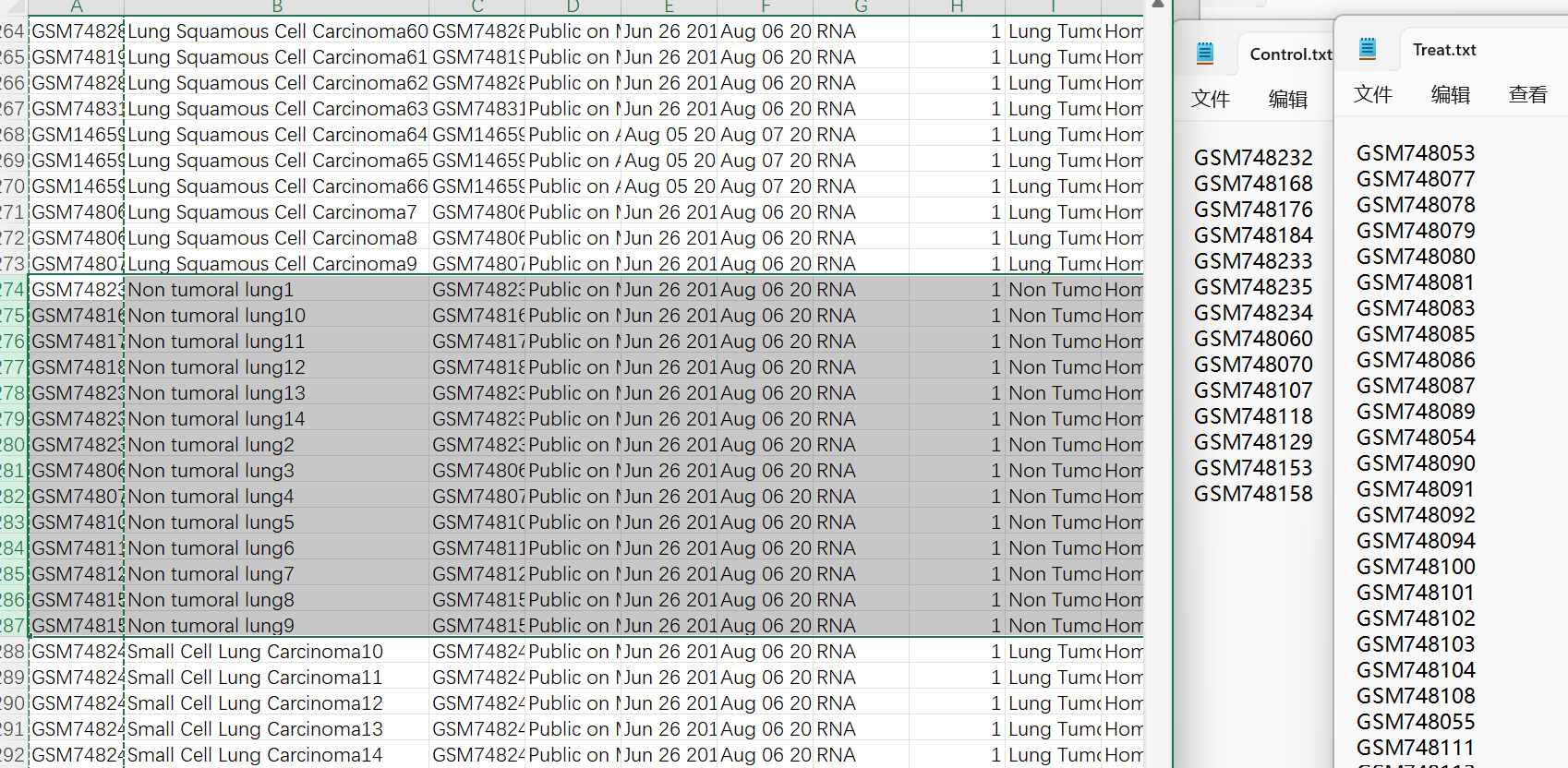
临床数据clinical_GSE30219.csv预处理(获得分组信息):
-
按
title列排序:选中该列,上边工具栏数据->排序->扩展选定区域->排序依据tilte列 -
将
title列值为Non tumoral lung的几行对应的样本名复制到Control.txt中,其它样本名复制到Treat.txt中
library(limma);
library(pheatmap);
library(stringr);
library(ggplot2);
library(ggVolcano);
读入表达量数据:
data <- read.table("data\\GSE30219\\GSE30219.txt", header = T, sep = "\t", check.names = F, row.names = 1);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames); # 转化为matrix
# data=data[rowMeans(data)>1,]; # 去除低表达的基因
# boxplot(data.frame(data),col="#4DBBD5");
data <- normalizeBetweenArrays(data); # 标准化
# boxplot(data.frame(data),col="#4DBBD5"); # 根据箱型图是否变平整查看结果是否标准化
# 保存数据
write.table(data.frame(ID = rownames(data), data), file = "save_data\\geo_normalize.txt", sep = "\t", quote = F, row.names = F);
读取分组信息(每个样本是正常还是肿瘤),并进行差异分析和差异基因筛选(这部分过程与TCGA的类似):
注意:表达矩阵已经取过log2,无需再取
# 分组信息文件
Control <- read.table("data\\GSE30219\\Control.txt", header = F, sep = "\t", check.names = F);
Treat <- read.table("data\\GSE30219\\Treat.txt", header = F, sep = "\t", check.names = F);
# 分组信息
conNum <- length(rownames(Control));
treatNum <- length(rownames(Treat));
Type <- c(rep(1, conNum), rep(2, treatNum));
# 将上面得到的表达矩阵按照正常、肿瘤组排序
data <- cbind(data[, Control[, 1]], data[, Treat[, 1]]);
# 差异分析
outTab <- data.frame();
for(i in row.names(data)){
rt <- data.frame(expression = data[i,], Type = Type);
wilcoxTest <- wilcox.test(expression ~ Type, data = rt);
pvalue <- wilcoxTest$p.value;
conGeneMeans <- mean(data[i, 1:conNum]);
treatGeneMeans <- mean(data[i,(conNum+1):ncol(data)]);
logFC <- treatGeneMeans-conGeneMeans;
conMed <- median(data[i, 1:conNum]);
treatMed <- median(data[i, (conNum+1):ncol(data)]);
diffMed <- treatMed-conMed;
if(((logFC>0) & (diffMed>0)) | ((logFC<0) & (diffMed<0))){
outTab <- rbind(
outTab,
cbind(
gene = i,
conMean = conGeneMeans,
treatMean = treatGeneMeans,
logFC = logFC,
pValue = pvalue
)
);
}
}
pValue <- outTab[,"pValue"];
fdr <- p.adjust(as.numeric(as.vector(pValue)), method = "fdr");
outTab <- cbind(outTab, fdr = fdr);
# 保存差异分析结果
write.table(outTab, file = "save_data\\all.Wilcoxon.txt", sep = "\t", row.names = F, quote = F);
# 差异基因筛选
logFCfilter <- 1;
fdrFilter <- 0.05;
# 保存差异基因筛选
outDiff <- outTab[(
abs(as.numeric(as.vector(outTab$logFC)))>logFCfilter &
as.numeric(as.vector(outTab$fdr))<fdrFilter
), ];
write.table(outDiff, file = "save_data\\diff.Wilcoxon.txt", sep = "\t", row.names = F, quote = F);
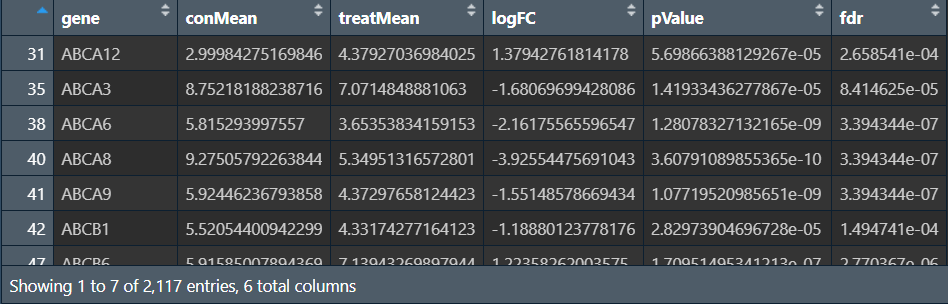
outTab:
outDiff:
画图:
# 热图
geneNum <- 50; # 要展示的基因数量
outDiff <- outDiff[order(as.numeric(as.vector(outDiff$logFC))),]; # 按logFC排序
diffGeneName <- as.vector(outDiff[, 1]); # 差异基因名
diffLength <- length(diffGeneName); # 差异基因长度
hmGene <- c(); # 展示的基因
if(diffLength>(2*geneNum)){
hmGene <- diffGeneName[c(1:geneNum, (diffLength-geneNum+1):diffLength)];
}else{
hmGene <- diffGeneName;
}
hmExp <- log2(data[hmGene, ]+0.01); # 取log2
Type <- c(rep("Normal", conNum),rep("Tumor", treatNum)); # 分组信息
names(Type) <- colnames(data);
Type <- as.data.frame(Type);
pdf(file = "save_data\\geo_heatmap.pdf", width = 10, height = 6.5);
pheatmap(
hmExp,
annotation = Type,
color = colorRampPalette(c(rep("#4DBBD5", 5), "white", rep("#E64B35", 5)))(50),
cluster_cols = F,
show_colnames = F,
scale = "row",
fontsize = 8,
fontsize_row = 5,
fontsize_col = 8
);
dev.off();
# 火山图
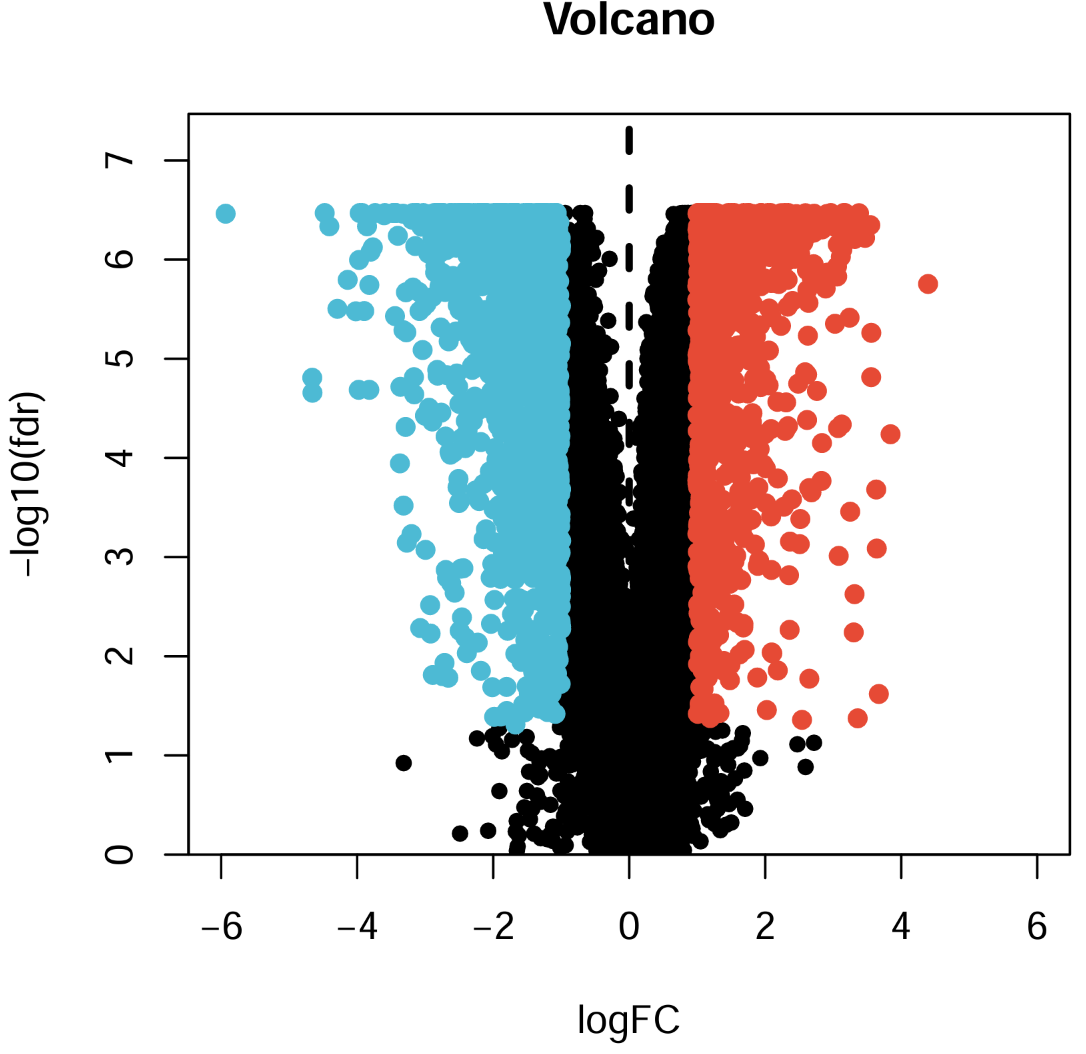
pdf(file = "save_data\\geo_vol.pdf", width = 5, height = 5);
xMax <- 6;
yMax <- max(-log10(outTab$fdr))+1;
plot(
as.numeric(as.vector(outTab$logFC)),
-log10(outTab$fdr),
xlab = "logFC",
ylab = "-log10(fdr)",
main = "Volcano",
xlim = c(-xMax, xMax),
ylim = c(0, yMax),
yaxs = "i",
pch = 20,
cex = 1.2
);
diffSub <- subset(
outTab, fdr<fdrFilter & as.numeric(as.vector(logFC))>logFCfilter
); # x=logFC_filter右边的点
points(
as.numeric(as.vector(diffSub$logFC)),
-log10(diffSub$fdr),
pch = 20,
col = "#E64B35",
cex = 1.5
);
diffSub <- subset(
outTab,
fdr<fdrFilter & as.numeric(as.vector(logFC))<(-logFCfilter)
); # x=-logFC_filter左边的点
points(
as.numeric(as.vector(diffSub$logFC)),
-log10(diffSub$fdr),
pch = 20,
col = "#4DBBD5",
cex = 1.5
);
abline(v = 0, lty = 2, lwd = 3); #分界线
dev.off();
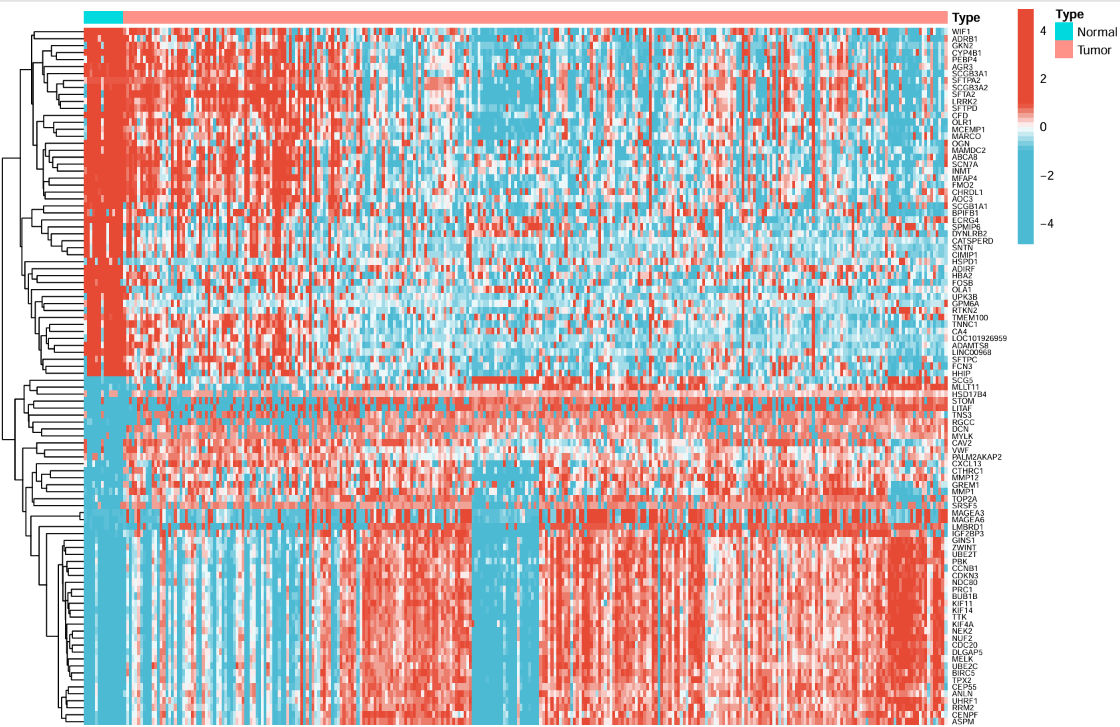
左面的树状线是基因聚类,右面(纵轴)是基因名称,横轴为不同的样本,每个点的颜色表示表达量大小,最上面的type标识每个样本是肿瘤/正常组