b站生信课程02-5
r-bioinfolessonb站生信课程TCGA及GEO数据挖掘入门必看P35-P43和P50笔记——GTEx和ICGC数据的下载与整理、泛癌差异基因箱线图、WGCNA、PCA、批量生存分析、自定义通路富集打分、IPS预测免疫治疗反应
写在前面:本篇教程来自b站课程TCGA及GEO数据挖掘入门必看 P35-P43和P50 相关资料下载
GTEx数据下载和整理
TCGA中正常组样本过少,差异分析时可能出现样本数不平衡的问题,因此考虑导入GTEx数据库的正常组织转录组测序数据
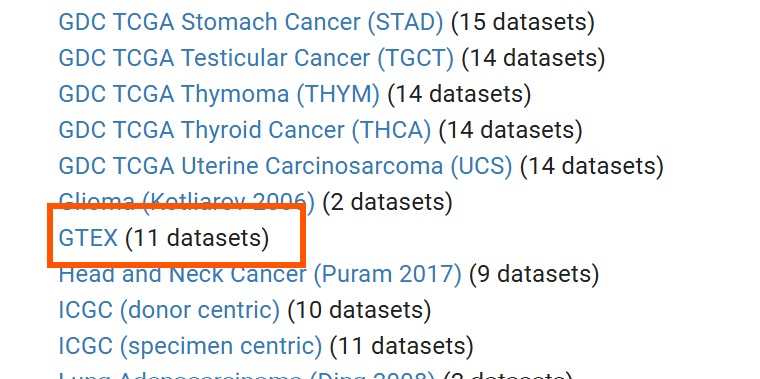
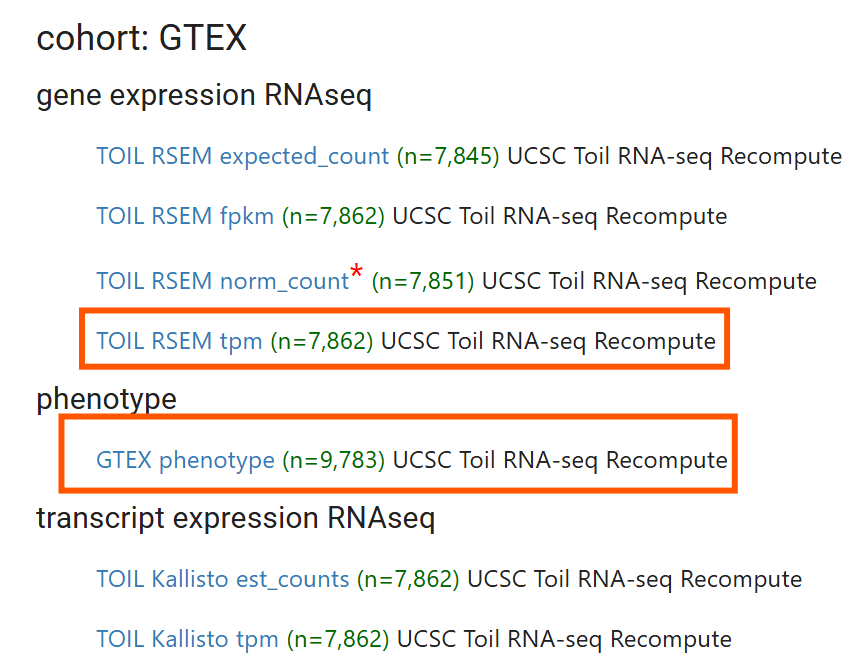
进入ucsc官网,点击Launch Xena,再点击上边栏的Data Sets,找到GTEX (11 datasets),点击进入
if(!require("AnnoProbe", quietly = T))
{
library(devtools);
devtools::install_github("jmzeng1314/AnnoProbe");
}
if(!require("tinyarray", quietly = T))
{
library(devtools);
devtools::install_github("xjsun1221/tinyarray", upgrade = F);
}
library(data.table);
library(tidyverse);
library(AnnoProbe);
library(tinyarray);
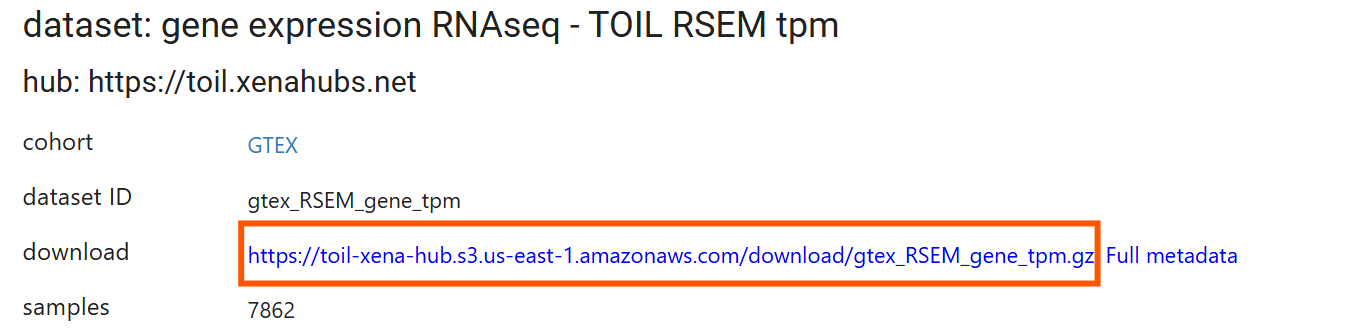
读取tpm表达矩阵:
dat <- data.table::fread("data\\GTEx\\gtex_RSEM_gene_tpm.gz", data.table = F); # 读入
exp <- column_to_rownames(dat, "sample") %>% # 将第一列sample转化行名
as.matrix();
rownames(exp) <- rownames(exp) %>%
str_split("\\.",simplify = T) %>% .[, 1]; # 删除行名`.`之后的字符
an <- annoGene(rownames(exp), ID_type = "ENSEMBL"); # 获取探针对应的symbol
exp <- trans_array(exp, ids = an, from = "ENSEMBL", to = "SYMBOL"); # 转换行名
View(exp[1:5, 1:5]):
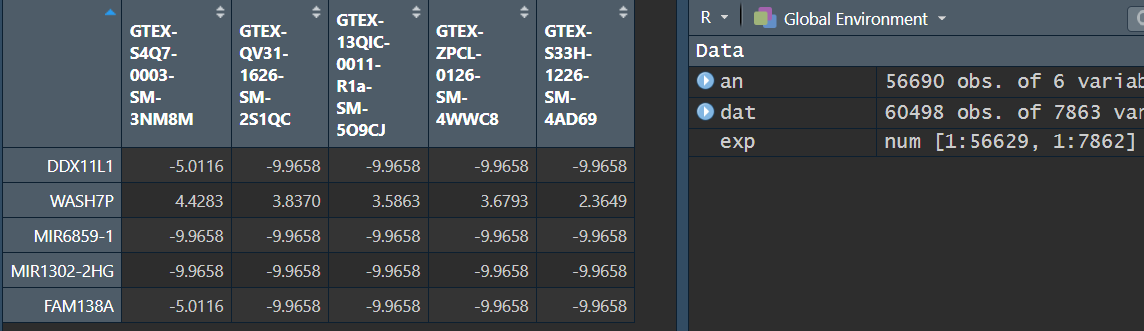
此时exp表达矩阵包含所有的样本
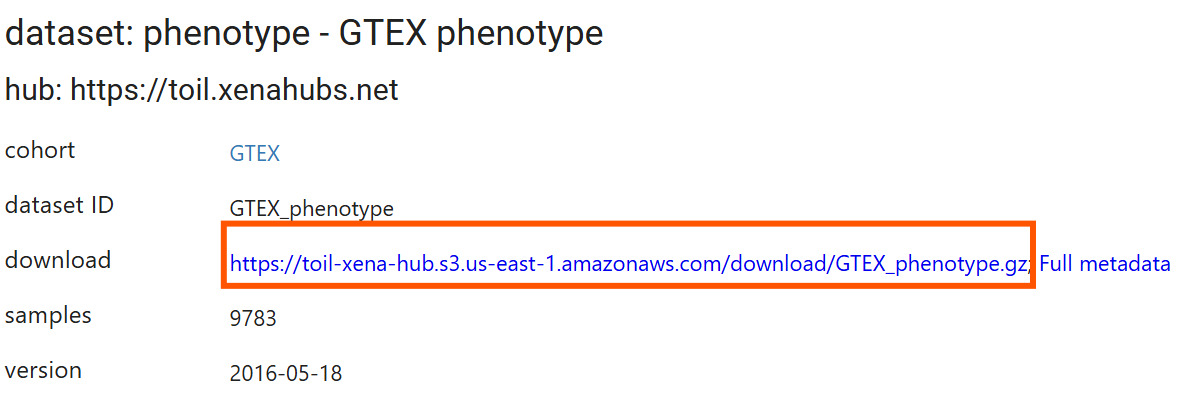
读取临床数据:
clinical <- data.table::fread("data\\GTEx\\GTEX_phenotype.gz"); # 读入
clinical <- clinical[clinical$`_primary_site`!="<not provided>", ]; # 删除未知补位的样本
colnames(clinical)[3] <- "site"; # 修改列名
clinical.subset <- subset(clinical, site=="Lung"); # 获取指定部位(肺部)的样本
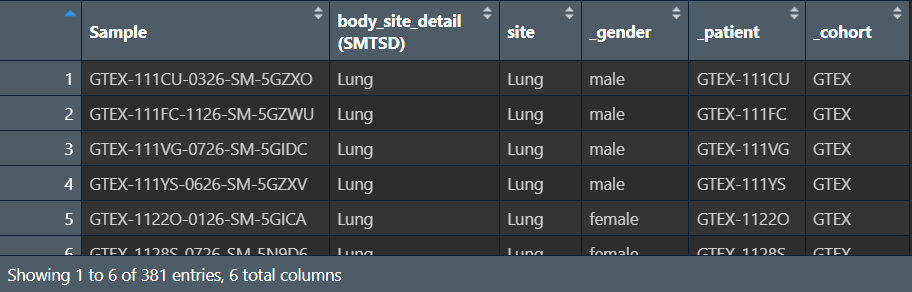
提取共同样本:即对exp表达矩阵进行筛选(肺部的样本)
s <- intersect(colnames(exp),clinical.subset$Sample); # 共同样本
clinical.subset = clinical.subset[match(s, clinical.subset$Sample), ];
exp <- exp[,s]; # 提取
# 因为XENA的数据是经过log2(tpm+0.001)处理的,需要转化回来
exp <- round(2^exp-0.001, 4); # 保留小数点后四位
# 保存数据
write.table(data.frame(ID = rownames(exp), exp), file = "save_data\\Lung.txt", sep = "\t", quote = F, row.names = F);
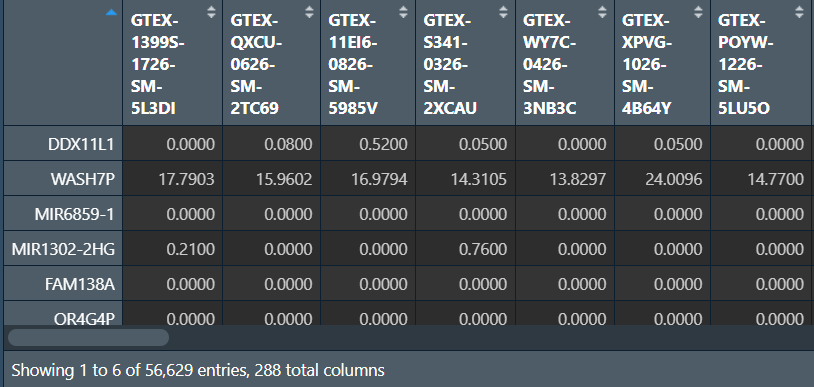
列名是样本名,行名是基因名,数据值是表达量
泛癌差异基因箱线图(线上)
进入TIMER官网,点击Gene_DE

在Gene Expression:中选择想要绘制的基因,之后点击Submit提交,即可绘图,点击PDF保存为pdf各式文件
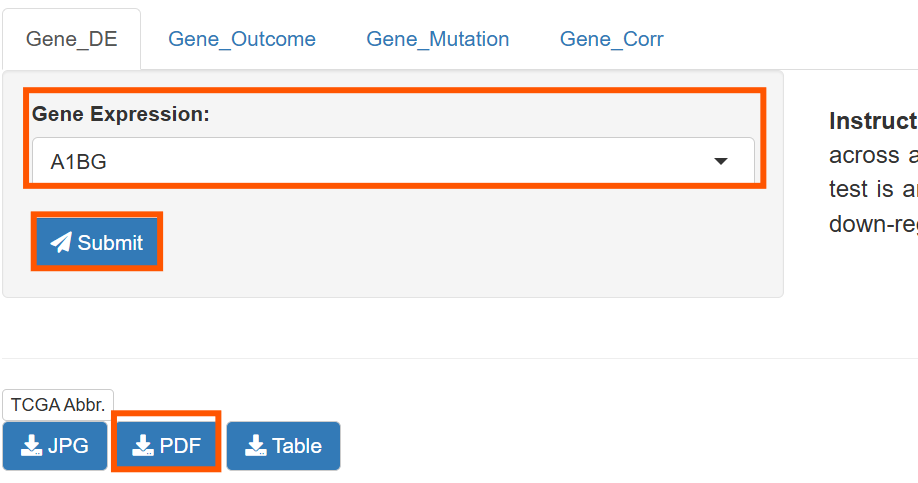
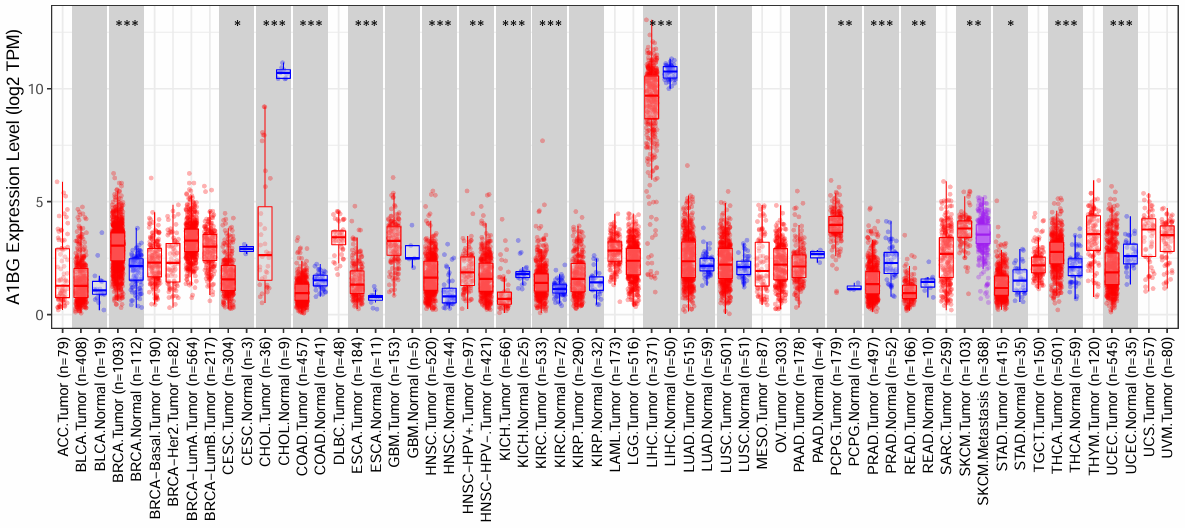
横坐标是来自TCGA不同肿瘤数据集的样本,纵坐标是指定基因的表达量(tpm取log2),红色是肿瘤样本、蓝色是正常样本、紫色是转移样本,上面的*就是表达量在肿瘤/正常/转移样本间的差异
当要研究单个基因,或者研究是聚焦于某一个基因进行时,可以通过这样的方式查看它泛癌的研究,对这个基因进行扩展
WGCNA
加权基因共表达网络分析(weighted correlation network analysis, WGCNA)是用来描述不同样本间基因关联模式的系统生物学方法,可以用来鉴定高度协同变化的基因集,并根据基因集的内连性和基因集与表型间关联鉴定候补生物标记基因或治疗靶点
总的来说,WGCNA可以鉴定协同变化的基因集合,并探究基因集合与表型间的关联
需要数据:tpm表达矩阵、临床信息(性别、生存状态、T分期,也可以为肿瘤/正常组,这里只取肿瘤组样本分析)
if(!require("impute", quietly = T))
{
library("BiocManager");
BiocManager::install("impute");
}
if(!require("fastcluster", quietly = T))
{
install.packages("fastcluster");
}
if(!require("dynamicTreeCut", quietly = T))
{
install.packages("dynamicTreeCut");
}
if(!require("WGCNA", quietly = T))
{
install.packages("WGCNA");
}
library(limma);
library(WGCNA);
读取tpm表达矩阵和临床数据
临床数据处理:行名是样本名
-
性别:
FEMALE->1MALE->2 -
生存状态:死亡用2表示,存活用1表示
-
T分期:用1、2、3、…表示
# 表达矩阵
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", header = T, sep = "\t", check.names = F,row.names = 1);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
# 仅保留肿瘤样本
group <- sapply(strsplit(colnames(data),"\\-"), "[", 4);
group <- sapply(strsplit(group, ""), "[", 1);
group <- gsub("2", "1", group);
data <- data[, group==0];
# 修改样本名
colnames(data) <- substr(colnames(data), 1, 12);
colnames(data) <- gsub('[.]', '-', colnames(data));
# 去除低表达的基因
data <- data[rowMeans(data)>0.5, ];
# 临床数据
library("readxl");
cli <- read_excel("save_data\\clinical.xlsx");
cli <- column_to_rownames(cli, "bcr_patient_barcode"); # 样本名为行名
cli <- cli[, c("gender", "vital_status", "T")]; # 只保留性别、生存状态、T分期列
colnames(cli) <- c("Sex", "State", "T"); # 改列名
cli$State <- ifelse(cli$State=='Alive', 1, 2); # 死亡用2表示,存活用1表示
cli$Sex <- ifelse(cli$Sex=='FEMALE', 1, 2); # FEMALE->1 MALE->2
cli$`T` <- substr(cli$`T`, 1, 1); # T列只取第一个字符
# 提取共有样本
sameSample <- intersect(row.names(cli), colnames(data));
cli <- cli[sameSample, ];
data <- data[, sameSample];
# 保存处理后的临床数据
write.table(data.frame(ID = rownames(cli), cli), "save_data\\WGCNA_cli.txt", sep = '\t', quote = F, row.names = F);
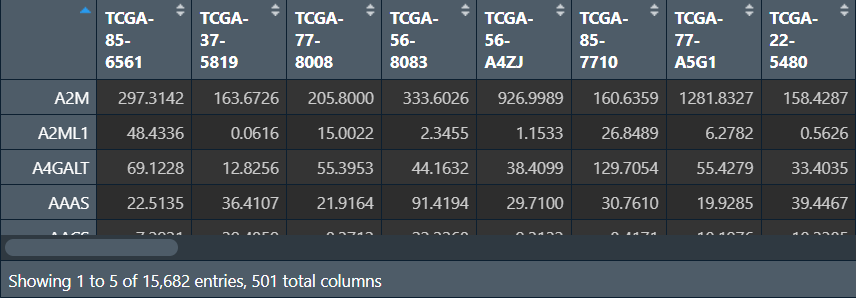
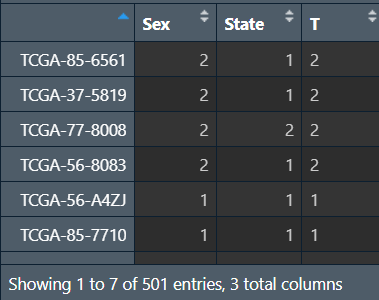
基因选取:选择波动最大的前30%的基因进行WGCNA分析
注:纳入的基因不易过多,否则需要运行很长一段时间,通常用5000个左右基因进行分析
selectGenes <- names(
tail(
sort(
apply(data, 1, sd)
),
n = round(nrow(data)*0.3)
)
);
data <- data[selectGenes, ];
datExpr0 <- t(data);
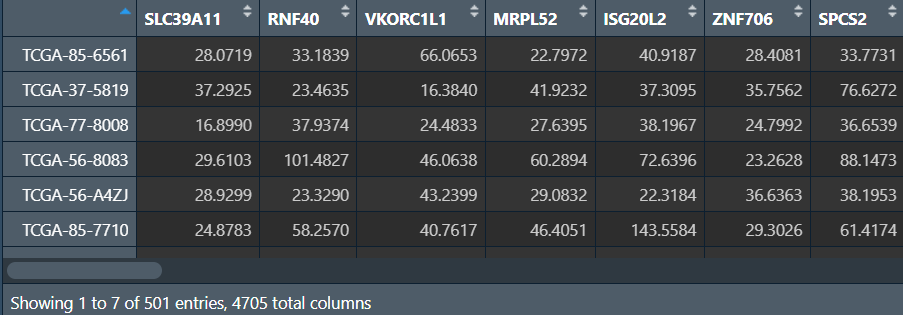
可以看到现在有4705个基因
检查缺失值和识别离群值(异常值):
gsg <- goodSamplesGenes(datExpr0, verbose = 3);
gsg$allOK; # 如果为true就是没有,不执行下面的代码
if (!gsg$allOK){
if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")));
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")));
datExpr0 <- datExpr0[gsg$goodSamples, gsg$goodGenes];
}
聚类所有样本,观察是否有离群值或异常值,并选取剪切值:
sampleTree <- hclust(dist(datExpr0), method = "average");
# 画图1
pdf(file = "save_data\\1_sample_cluster.1.pdf", width = 12, height = 9);
par(cex = 0.6);
par(mar = c(0,4,2,0));
plot(
sampleTree,
main = "Sample clustering to detect outliers", sub = "", xlab = "",
cex.lab = 1.5, cex.axis = 1.5, cex.main = 2
);
dev.off();

红框中的样本聚类得比较好,蓝框为离群值
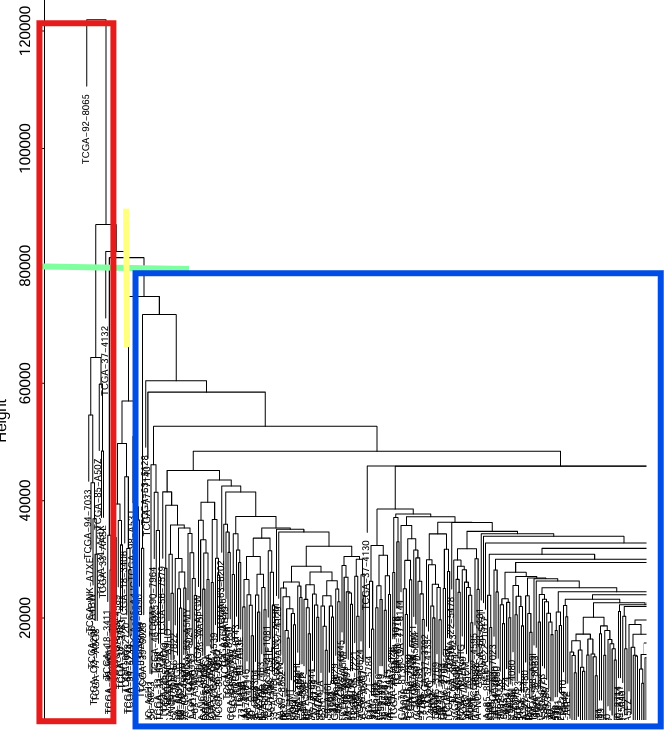
剪切值:当剪切值设为80000时,以树干(黄色线)为分界线,树状图左边的分支(红框)会被删除,右边(蓝框)保留
这里选取剪切值为100000,如果不想要删除可以将cutHeight设置的高些
cutHeight <- 100000; # 剪切值
# 画图2(剪切线)
pdf(file = "save_data\\1_sample_cluster.2.pdf", width = 12, height = 9);
par(cex = 0.6);
par(mar = c(0,4,2,0));
plot(
sampleTree,
main = "Sample clustering to detect outliers", sub = "", xlab = "",
cex.lab = 1.5, cex.axis = 1.5, cex.main = 2
);
abline(h = cutHeight, col = "red");
dev.off();
# 剪切
clust <- cutreeStatic(sampleTree, cutHeight = cutHeight, minSize = 10);
keepSamples <- (clust==1);
datExpr0 <- datExpr0[keepSamples, ];
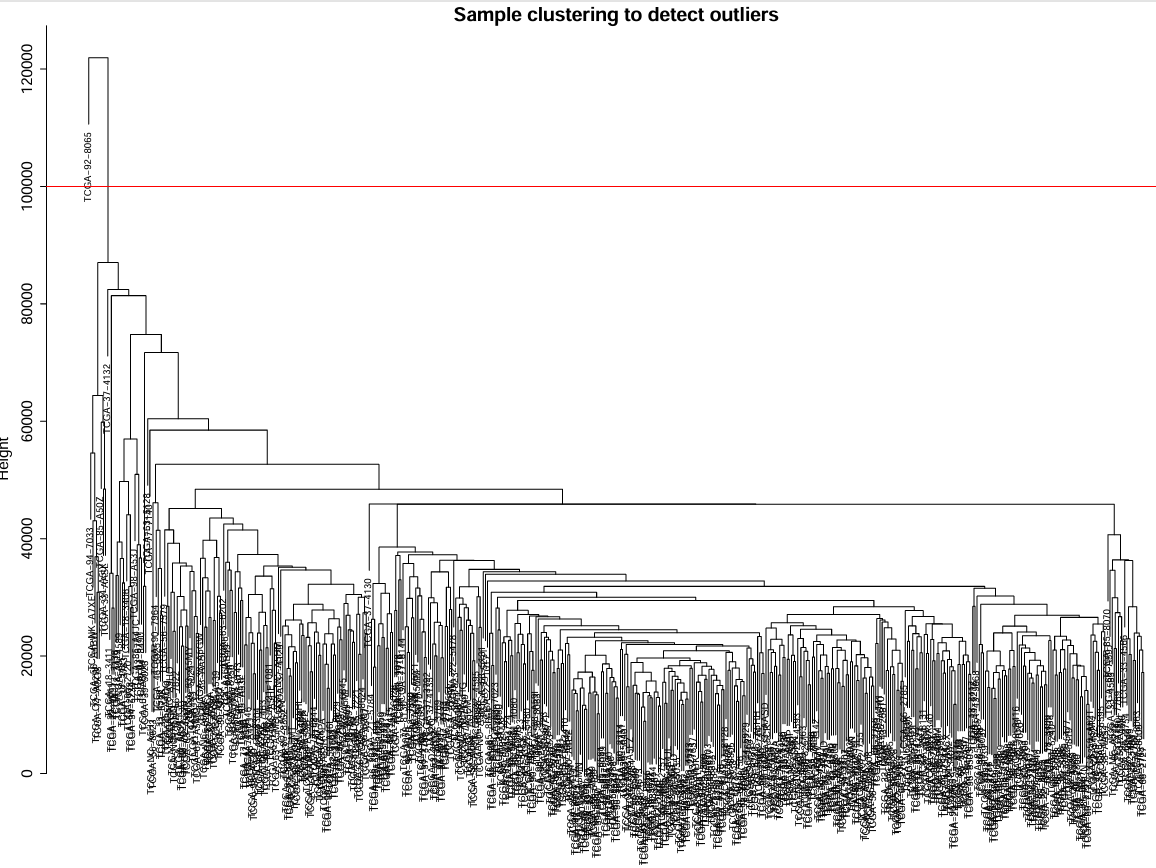
可以看到原来是501个样本,现在是500个样本
构建自动化网络和检测模块,绘制power值散点图,选择软阈值:
enableWGCNAThreads(); # 多线程工作
powers <- c(1:20); # 幂指数范围1:20
sft <- pickSoftThreshold(datExpr0, powerVector = powers, verbose = 5);
pdf(file = "save_data\\2_scale_independence.pdf", width = 9, height = 5);
par(mfrow = c(1, 2));
cex1 <- 0.9; # 可以修改,如改成0.8
# 拟合指数与power值散点图,无标度拓扑拟合指数
plot(
sft$fitIndices[, 1],
-sign(sft$fitIndices[,3])*sft$fitIndices[, 2],
xlab = "Soft Threshold (power)", ylab = "Scale Free Topology Model Fit,signed R^2",
type = "n",
main = paste("Scale independence")
);
text(
sft$fitIndices[, 1],
-sign(sft$fitIndices[, 3])*sft$fitIndices[, 2],
labels = powers,
cex = cex1,col = "red"
);
abline(h = cex1, col = "red");
# 平均连通性与power值散点图
plot(
sft$fitIndices[,1],
sft$fitIndices[,5],
xlab = "Soft Threshold (power)", ylab = "Mean Connectivity",
type = "n",
main = paste("Mean connectivity")
);
text(
sft$fitIndices[, 1],
sft$fitIndices[, 5],
labels = powers,
cex = cex1, col = "red"
);
dev.off();
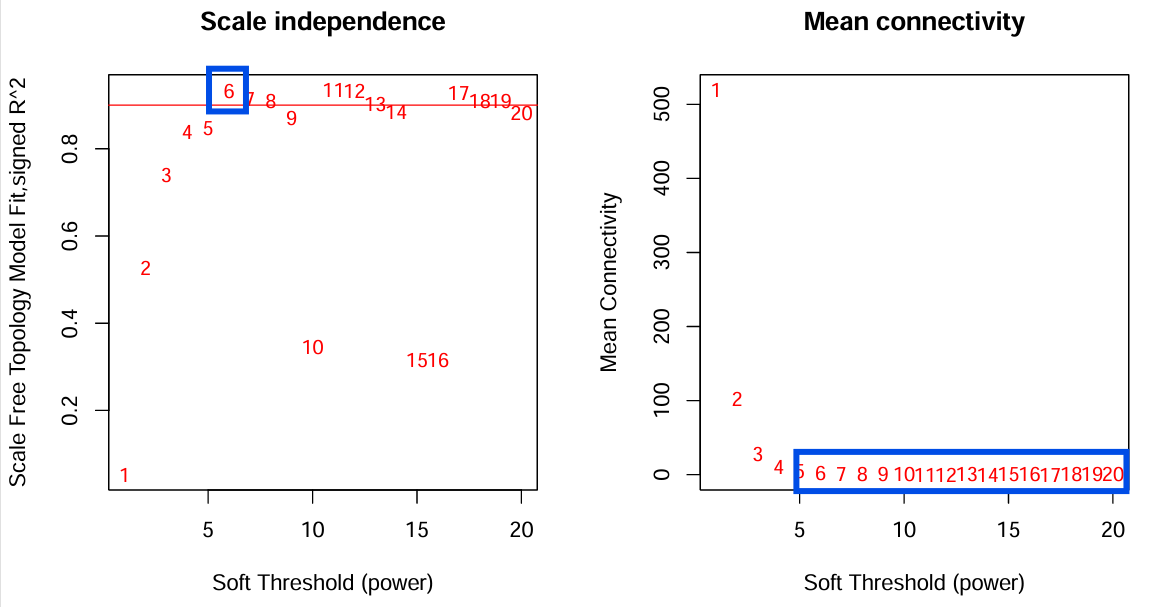
对于Scale independence,一般选择在0.9以上的,第一个达到0.9以上的数值();对于Mean connectivity,要选取较平滑的值
邻接矩阵转换,查看最佳power值:
注:如果显示的结果为 NA,则表明系统无法给出合适的软阈值,这时候就需要自己挑选软阈值
softPower <- sft$powerEstimate;
adjacency <- adjacency(datExpr0, power = softPower);
softPower;
# 如无合适软阈值时,可以按以下条件选择
if (is.na(softPower)){
softPower <- ifelse(
nSamples<20,
ifelse(type == "unsigned", 9, 18),
ifelse(
nSamples<30,
ifelse(type == "unsigned", 8, 16),
ifelse(
nSamples<40,
ifelse(type == "unsigned", 7, 14),
ifelse(type == "unsigned", 6, 12)
)
)
);
}
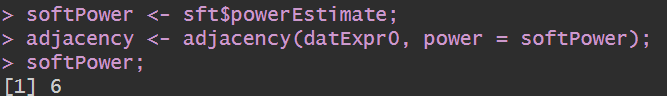
可以看到应选取的power值为6
基因聚类:
# TOM矩阵
TOM <- TOMsimilarity(adjacency);
dissTOM <- 1-TOM;
# 基因聚类
geneTree <- hclust(as.dist(dissTOM), method = "average");
pdf(file = "save_data\\3_gene_clustering.pdf", width = 12, height = 9);
plot(
geneTree,
xlab = "", sub = "", main = "Gene clustering on TOM-based dissimilarity",
labels = FALSE,
hang = 0.04
);
dev.off();
动态剪切模块识别:
#模块基因数目
minModuleSize <- 100; # 最小单个模块包含基因数
dynamicMods <- cutreeDynamic(
dendro = geneTree,
distM = dissTOM,
deepSplit = 2,
pamRespectsDendro = FALSE,
minClusterSize = minModuleSize
);
dynamicColors <- labels2colors(dynamicMods);
pdf(file = "save_data\\4_Dynamic_Tree.pdf", width = 8, height = 6);
plotDendroAndColors(
geneTree,
dynamicColors,
"Dynamic Tree Cut",
dendroLabels = FALSE,
hang = 0.03,
addGuide = TRUE,
guideHang = 0.05,
main = "Gene dendrogram and module colors"
);
dev.off();
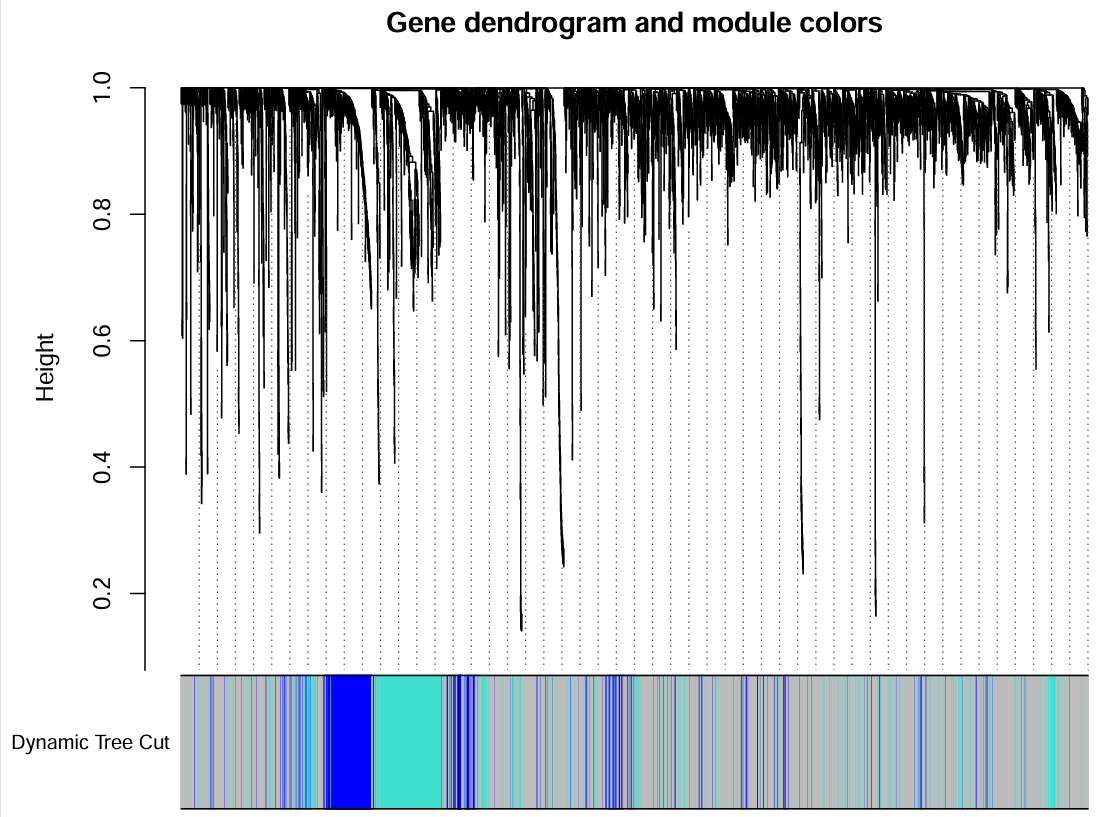
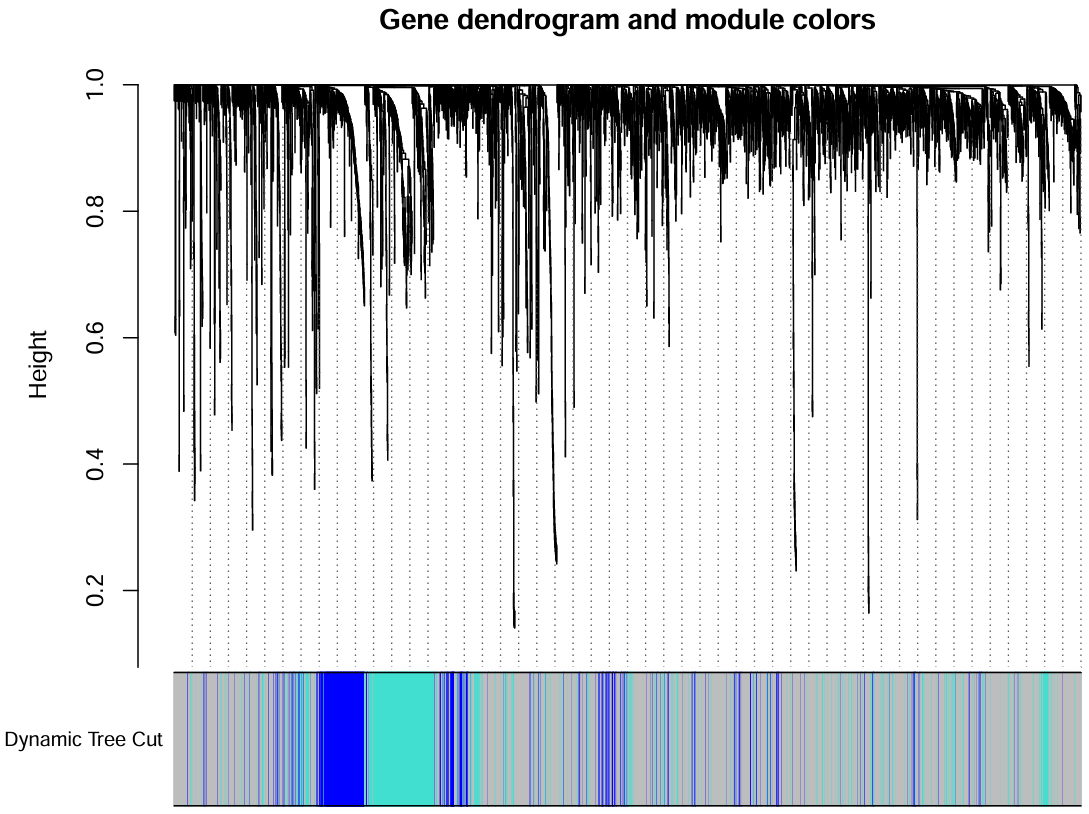
上面图是基因聚类的结果,下面是根据minModuleSize划分的不同的模块,每种颜色代表一个模块
查找相似模块:通过计算模块的代表性模式和模块之间的定量相似性评估,合并表达图谱相似的模块
MEList <- moduleEigengenes(datExpr0, colors = dynamicColors);
MEs <- MEList$eigengenes;
MEDiss <- 1-cor(MEs);
METree <- hclust(as.dist(MEDiss), method = "average");
pdf(file = "save_data\\5_Clustering_module.pdf", width = 7, height = 7);
plot(
METree,
main = "Clustering of module eigengenes", xlab = "", sub = ""
);
MEDissThres <- 0.3; # 剪切高度,可修改,表示将高度<0.3的模块合并
abline(h = MEDissThres, col = "red");
dev.off();
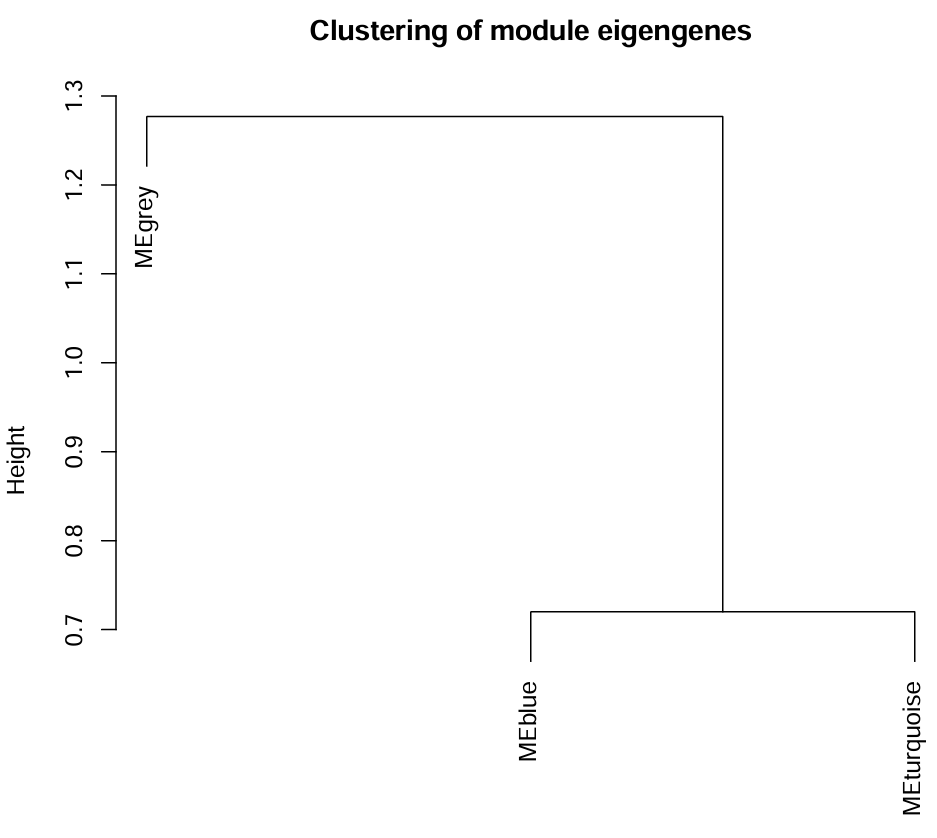
可以看到有三个模块,它们的高度都>0.7,没有<0.3的模块,可不用进行下面的相似模块合并(执行一遍下面的代码也可以)
相似模块合并:
merge <- mergeCloseModules(
datExpr0,
dynamicColors,
cutHeight = MEDissThres,
verbose = 3
);
mergedColors <- merge$colors;
mergedMEs <- merge$newMEs;
pdf(file = "save_data\\6_merged_dynamic.pdf", width = 8, height = 6);
plotDendroAndColors(
geneTree,
mergedColors,
"Dynamic Tree Cut",
dendroLabels = FALSE,
hang = 0.03,
addGuide = TRUE,
guideHang = 0.05,
main = "Gene dendrogram and module colors"
);
dev.off();
moduleColors <- mergedColors;
table(moduleColors);
colorOrder <- c("grey", standardColors(50));
moduleLabels <- match(moduleColors, colorOrder)-1;
MEs <- mergedMEs;
MEs:
提取共有样本:
# 重新读入临床数据
cli2 <- read.table("save_data\\WGCNA_cli.txt", header = T, sep = "\t", check.names = F, row.names = 1);
# 提取共有样本
sameSample2 <- intersect(row.names(cli2), rownames(MEs));
MEs <- MEs[sameSample2,];
datTraits <- cli2[sameSample2, ];
nGenes <- ncol(datExpr0);
nSamples <- nrow(datExpr0);
# 相关性分析
moduleTraitCor <- cor(MEs, datTraits, use = "p");
moduleTraitPvalue <- corPvalueStudent(moduleTraitCor, nSamples);
pdf(file = "save_data\\7_Module_trait.pdf", width = 6.5, height = 5.5);
textMatrix <- paste( # 不能用paste0
signif(moduleTraitCor, 2),
"\n(", signif(moduleTraitPvalue, 1), ")",
sep = ""
);
dim(textMatrix) = dim(moduleTraitCor);
par(mar = c(5, 10, 3, 3));
labeledHeatmap(
Matrix = moduleTraitCor,
xLabels = names(datTraits),
yLabels = names(MEs),
ySymbols = names(MEs),
colorLabels = FALSE,
colors = blueWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 0.5,
zlim = c(-1,1),
main = "Module-trait relationships"
);
dev.off();
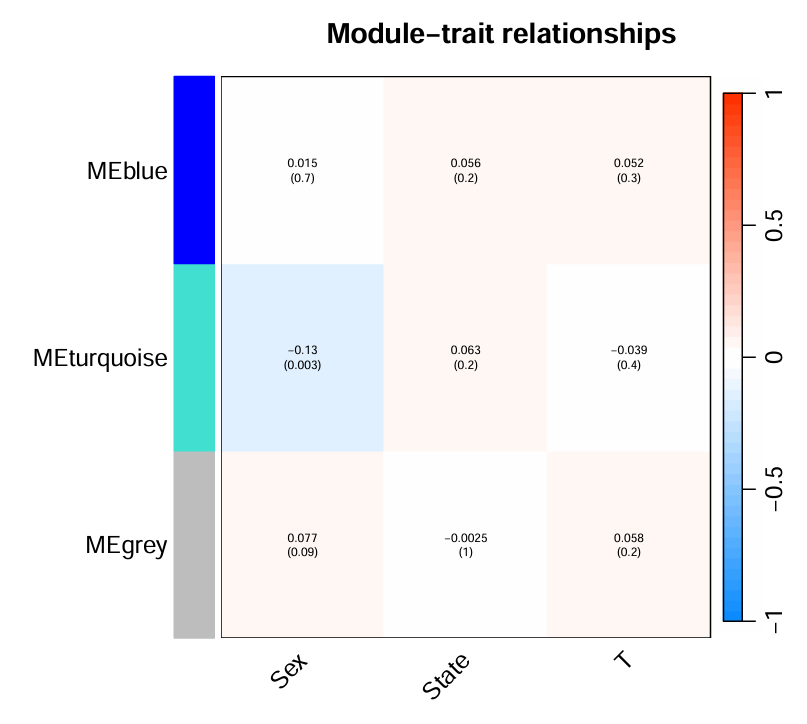
横坐标是表型/临床特征(性别、生存状态、临床T分期),纵坐标是分析模块(将基因分成了蓝色、青色、灰色三个模块),3*3个数据值:红色表示正相关、蓝色负相关、0不相关,方块中上面的数字表示相关性系数,下面的括号里的表示相关性系数对应的p值
标识每个基因所在的模块,并保存每个模块的基因:
# 每个基因所在的模块
probes <- colnames(datExpr0);
geneInfo0 <- data.frame(
probes = probes,
moduleColor = moduleColors
);
geneOrder <- order(geneInfo0$moduleColor);
geneInfo <- geneInfo0[geneOrder, ];
write.table(geneInfo, file = "save_data\\module_all.txt", sep = "\t", row.names = F, quote = F);
# 每个模块的基因
for (mod in 1:nrow(table(moduleColors))){
modules <- names(table(moduleColors))[mod];
probes <- colnames(datExpr0);
inModule <- (moduleColors==modules);
modGenes <- probes[inModule];
write.table(
modGenes,
file = paste0("save_data\\module_", modules, ".txt"),
sep = "\t", row.names = F, col.names = F, quote = F
);
}
geneInfo:
View(as.data.frame(modGenes)):(共有三个,这里以其中为1个为例)
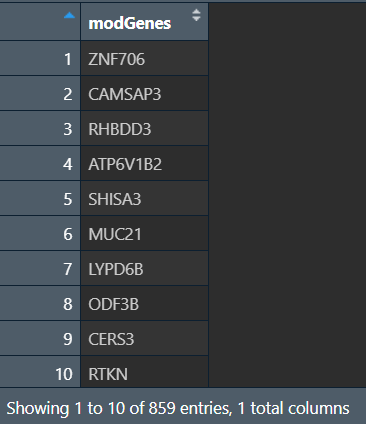
计算GS和MM:
-
GS(重要性):所有基因表达谱与这个模块的基因的相关性(cor),其值代表这个基因与模块之间的关系。这个值的绝对值接近0,这个基因就越不是这个模块的一部分;这个值的绝对值接近1,这个基因就越与这个模块相关
-
MM(相关性):基因和表型性状之间的相关性的绝对值
以MEturquoise(青色的那个组)中的性别为例(因为在上面的相关图中,只有这个块的p值<0.05)
module <- "turquoise";
Selectedclinical <- "Sex";
Selectedclinical2 <- "Sex";
Selectedclinical <- as.data.frame(datTraits[, Selectedclinical]);
names(Selectedclinical) <- "Selectedclinical";
modNames <- substring(names(MEs), 3);
datExpr1 <- datExpr0[rownames(MEs), ];
geneModuleMembership <- as.data.frame(cor(datExpr1, MEs, use = "p"));
MMPvalue <- as.data.frame(corPvalueStudent(as.matrix(geneModuleMembership), nSamples));
names(geneModuleMembership) <- paste("MM", modNames, sep = "");
names(MMPvalue) <- paste("p.MM", modNames, sep = "");
geneTraitSignificance <- as.data.frame(cor(datExpr1, Selectedclinical, use = "p"));
GSPvalue <- as.data.frame(corPvalueStudent(as.matrix(geneTraitSignificance), nSamples));
names(geneTraitSignificance) <- paste("GS.", names(Selectedclinical), sep = "");
names(GSPvalue) <- paste("p.GS.", names(Selectedclinical), sep = "");
#画图
column <- match(module, modNames);
moduleGenes <- moduleColors==module;
outPdf <- paste("save_data\\", Selectedclinical2, "_", module, ".pdf", sep = "");
pdf(file = outPdf, width = 7, height = 7);
verboseScatterplot(
abs(geneModuleMembership[moduleGenes, column]),
abs(geneTraitSignificance[moduleGenes, 1]),
xlab = paste("Module Membership in", module, "module"),
ylab = paste("Gene significance for ", Selectedclinical2, sep = ""),
main = "Module membership vs. gene significance\n",
cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2,
col = module
);
dev.off();
Sex_turquoise.pdf:
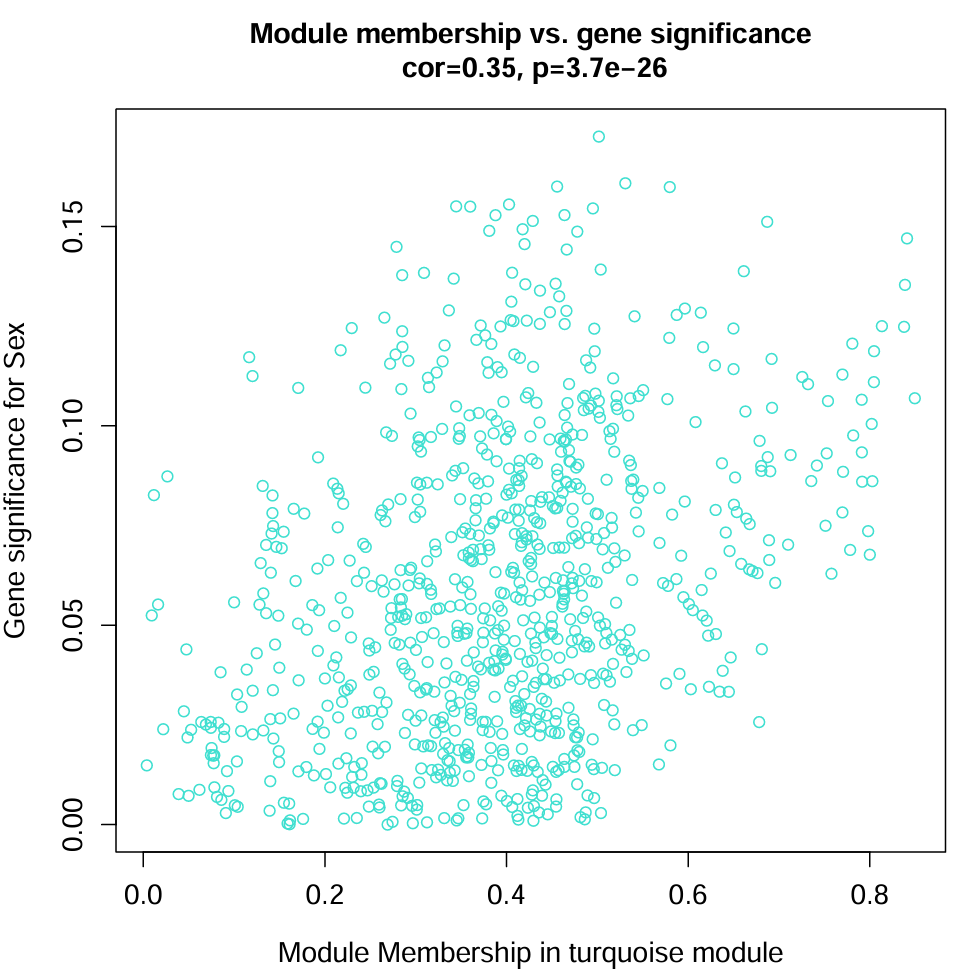
横坐标是MM在青色模块中的值,纵坐标是GS在性别中的值,副标题标识了相关性系数和p值
计算模块中的核心基因:
# 合并
datMM <- cbind(
geneModuleMembership[, paste("MM", module, sep = "")],
geneTraitSignificance
);
colnames(datMM)[1] <- paste("MM", module, sep="");
# datMM = datMM[moduleColors==module, ]; # 限定在特定模块中的基因
# 重要性和相关性阈值,调高以减少结果数量
geneSigFilter <- 0.15;
moduleSigFilter <- 0.4;
# 筛选
datMM <- datMM[abs(datMM[, ncol(datMM)])>geneSigFilter, ];
datMM <- datMM[abs(datMM[, 1])>moduleSigFilter, ];
# 导出
write.table(
row.names(datMM),
file = paste0("save_data\\hubGenes", module, ".txt"),
sep = "\t", row.names = F, col.names = F, quote = F
);
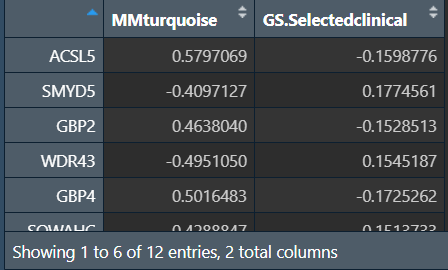
得到在青色模块中与性别相关的关键基因
PCA
主成分分析方法(Principal Component Analysis, PCA)是一种广泛使用的数据降维算法。主要思想是将n维特征映射到k维上,这k维是全新的正交特征,也被称为主成分,是在原有n维特征的基础上重新构造处理的k维特征
比如我们有一个数据集包含10000个基因,10000个基因数量太多,不好理解,可以通过PCA将这10000个基因转化为带有权重的几十个基因集合;PCA结果中越前面的基因,所占的权重越高,因此画图时通常取前2或3个基因集合,用于代替整个表达模式
需要数据:各样本的差异基因表达量和风险分组
if(!require("scatterplot3d", quietly = T))
{
install.packages("scatterplot3d");
}
library(limma);
library(ggplot2);
library(scatterplot3d);
读取数据:
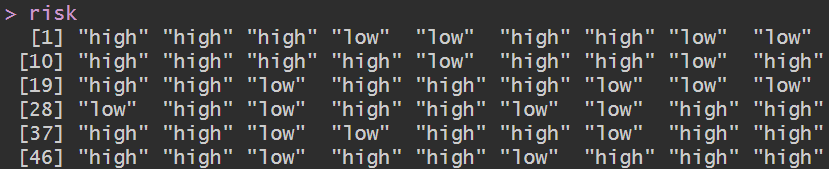
rt <- read.table("save_data\\risk.txt", header = T, sep = "\t", check.names = F, row.names = 1);
risk <- as.vector(rt$risk); # 高/低风险组
data <- rt[, 3:(ncol(rt)-2)]; # 各样本的各基因表达量
PCA分析并绘图:
# PCA分析
data.pca <- prcomp(data, scale. = TRUE);
pcaPredict <- predict(data.pca);
# 绘图数据(2d图)
PCA <- data.frame(
PC1 = pcaPredict[, 1],
PC2 = pcaPredict[, 2],
risk = risk
);
PCA.mean <- aggregate(PCA[, 1:2], list(risk = PCA$risk), mean);
# 2d图
pdf(file = "save_data\\PCA.2d.pdf", width = 5.5, height = 4.75);
ggplot(data = PCA, aes(PC1, PC2)) +
geom_point(aes(color = risk, shape = risk)) +
scale_colour_manual(
name = "risk",
values = c("DarkOrchid", "Orange2")
) +
theme_bw() +
labs(title = "PCA") +
theme(
plot.margin = unit(rep(1.5, 4), 'lines'),
plot.title = element_text(hjust = 0.5)
) +
annotate(
"text",
x = PCA.mean$PC1,
y = PCA.mean$PC2,
label = PCA.mean$risk,
cex = 7
) +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank()
);
dev.off();
# 3d图
color <- ifelse(risk=="high", "DarkOrchid", "Orange2");
pdf(file = "save_data\\PCA.3d.pdf", width = 7, height = 7);
par(oma = c(1, 1, 2.5, 1));
s3d <- scatterplot3d(
pcaPredict[, 1:3],
pch = 16,
color = color,
angle = 60
);
legend(
"top",
legend = c("High risk", "Low risk"),
pch = 16,
inset = -0.2,
box.col = "white",
xpd = TRUE,
horiz = TRUE,
col = c("DarkOrchid", "Orange2")
);
dev.off();
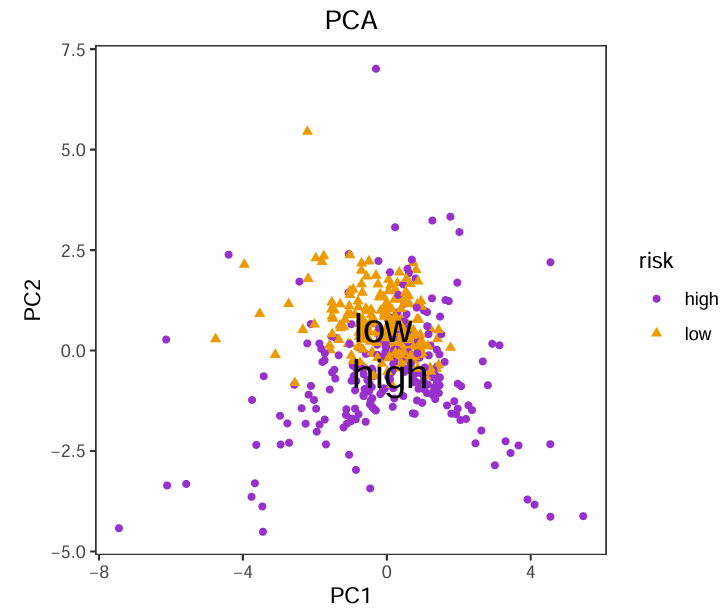
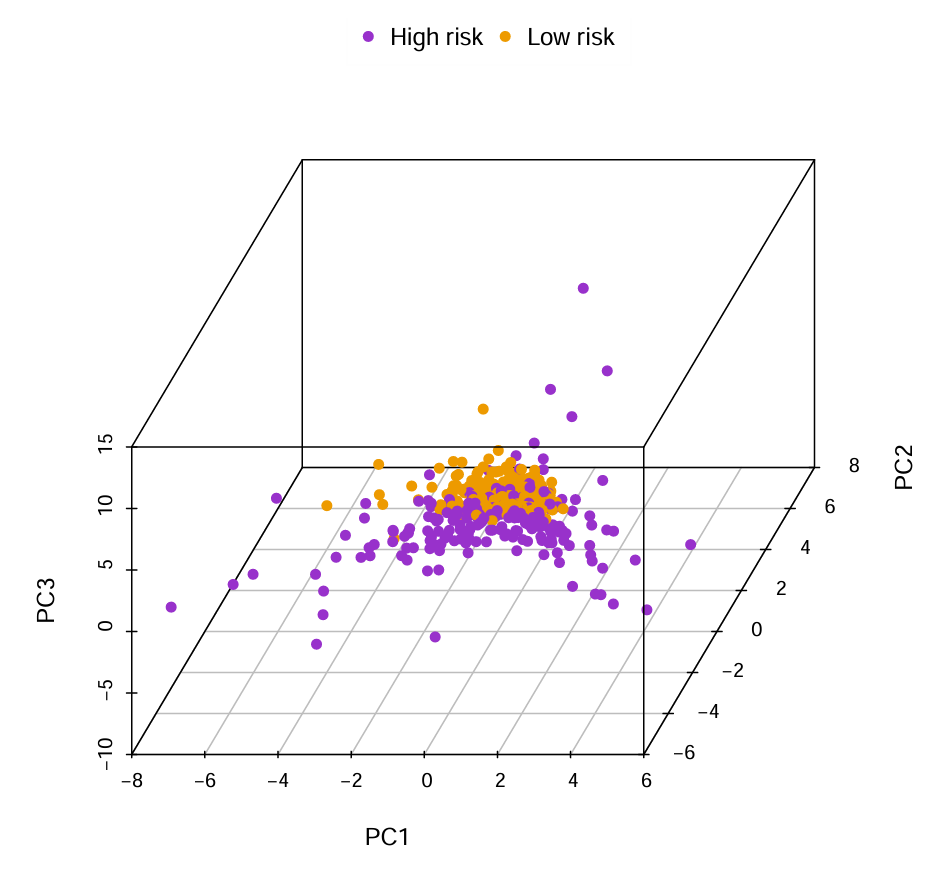
二维图横纵坐标分别是PC1和PC2(基因集合),不同的颜色代表不同的分组,可以看到低风险组主要位于左上角、高风险组主要位于右下角,表明高/低风险组的基因表达模式是不同的,三维图同理
批量生存分析
需要数据:tpm表达矩阵、生存信息(样本名、生存时间、状态)
library(limma);
library(survival);
library(survminer);
读取tpm表达矩阵、生存信息,合并:
# 表达矩阵
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", header = T, sep = "\t", check.names = F,row.names = 1);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
# 去除低表达的基因
data <- data[rowMeans(data)>1, ];
# 仅保留肿瘤样本
group <- sapply(strsplit(colnames(data),"\\-"), "[", 4);
group <- sapply(strsplit(group, ""), "[", 1);
group <- gsub("2", "1", group);
data <- data[, group==0];
# 修改样本名
colnames(data) <- substr(colnames(data), 1, 12);
colnames(data) <- gsub('[.]', '-', colnames(data));
# 转置
data <- t(data);
# 临床数据
cli <- read.table("save_data\\time_LUSC.txt", header = T, sep = "\t", check.names = F, row.names = 1);
cli$time <- cli$time/365;
# 合并
sameSample <- intersect(row.names(data), row.names(cli));
data <- data[sameSample, ];
cli <- cli[sameSample, ];
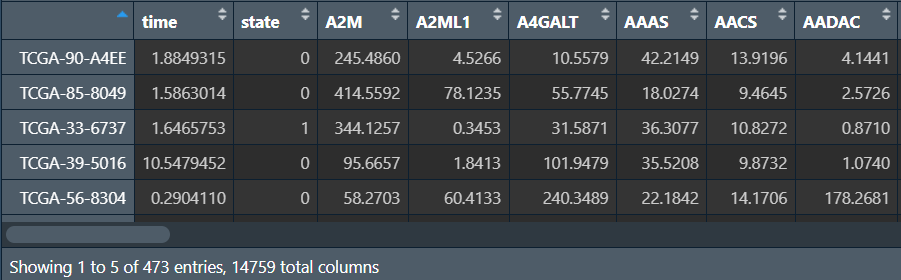
rt <- cbind(cli, data);
批量生存分析:
geneNum <- 50; # 为节省时间,仅判断前50个基因
outTab <- data.frame(); # 结果矩阵
for(gene in colnames(rt[, 3:(2+geneNum)])){
a <- rt[, gene] <= median(rt[, gene]); # 以中值为分界线,分成两组
diff <- survdiff(Surv(time, state) ~ a, data = rt);
pValue <- 1-pchisq(diff$chisq, df = 1); # p值
fit <- survfit(Surv(time, state) ~ a, data = rt);
if(pValue<0.05){ # 设置输出图片的p值阈值为0.05
outTab <- rbind(outTab, cbind(gene = gene, pval = pValue));
# 绘制生存曲线
if(pValue<0.001){ # 调整p值书写方式
pValue <- "<0.001";
}else{
pValue <- paste0("p=", sprintf("%0.3f", pValue)); # 保留三位小数
}
surPlot <- ggsurvplot(
fit,
data = rt,
conf.int = TRUE,
pval = pValue,
pval.size = 6,
legend.labs = c("High", "Low"),
legend.title = gene,
xlab = "Time(years)",
legend = c(0.8, 0.8),
font.legend = 10,
break.time.by = 1,
risk.table.title = "",
palette = c("Firebrick3","MediumSeaGreen"),
surv.median.line = "hv",
risk.table = T,
cumevents = F,
risk.table.height = .25
);
pdf(file = paste0("save_data\\sur.", gene, ".pdf"), width = 7, height = 6.5, onefile = FALSE);
print(surPlot);
dev.off();
}
}
# 保存基因其对应和p值
write.table(outTab, file = "save_data\\surv_result.txt", sep = "\t", row.names = F, quote = F);
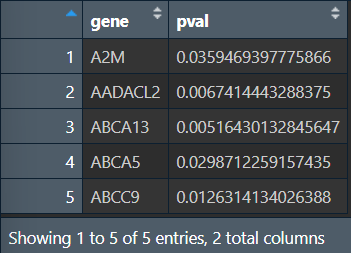
可以看到在前50个基因中,共筛选出5个生存时间在高/低表达组有显著差异的基因
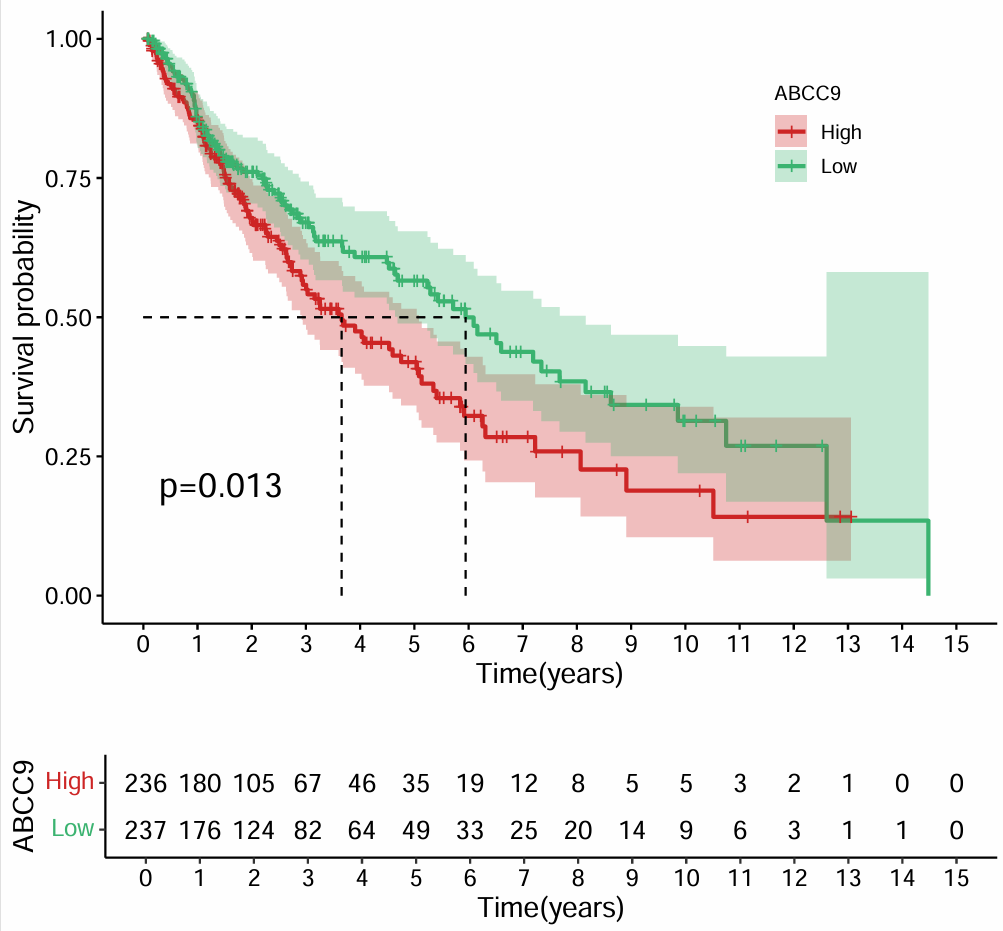
横坐标是生存时间/年,纵坐标代表生存的可能性(对应的时间有多少人存活),对于这个基因,高表达组的生存率低于低表达组
ICGC数据下载和整理
这里我们没有从ICGC官网下载,而是从ucsc数据下载,因为官网下载很慢,而且数据集需要多次处理(处理过程中对电脑内存要求很高)
进入ucsc官网
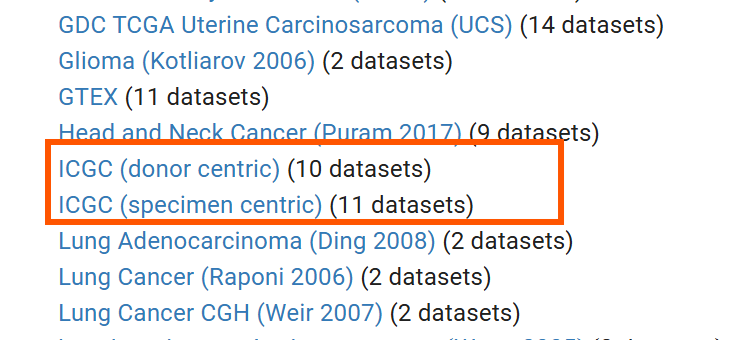
可以看到有两个ICGC数据集:
-
ICGC (donor centric)每条数据是一个病人 -
ICGC (specimen centric)每条数据是一个样本,因为一个病人可能有多条样本,所以这里面的数据条数更多
这里我们下载ICGC (specimen centric)的
-
US projects基因表达数据(RNAseq)
-
overall survival临床数据
-
specimen phenotype标识样本来源(来自哪个project/数据集)的数据
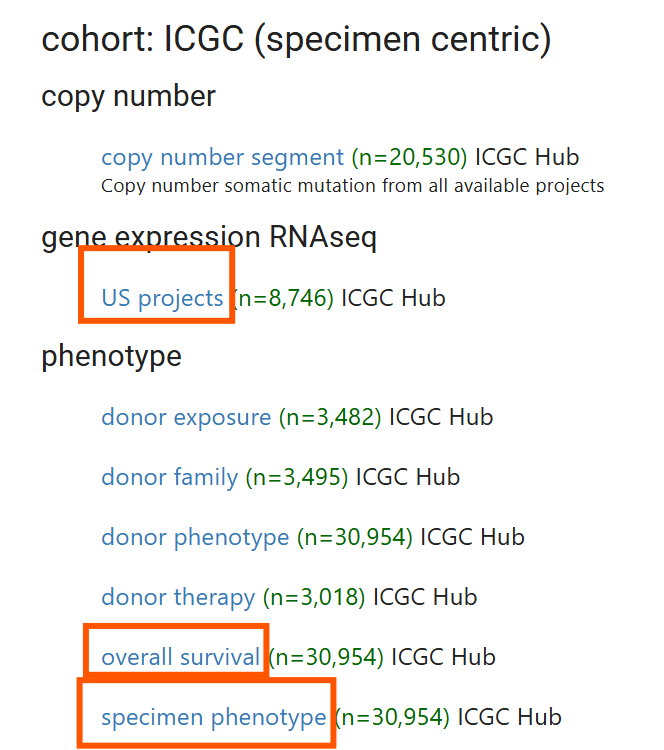
注:因为有些样本只有临床数据,没有基因表达数据,所以基因表达数据数量少于另两个
下载完成后解压成tsv文件:
-
donor.all_projects.overallSurvival_transfer_specimen列名为样本编号、病人编号、生存状态、生存时间 -
exp_seq.all_projects.specimen.USonly.xena.tsv列名为样本名,行名是基因名,数据是表达量 -
specimen.all_projects.tsv主要提供每个样本在哪个数据集,以及属于正常/肿瘤组
表达数据
library(tidyverse);
读取表达矩阵和分组信息:
# 表达矩阵
exp <- read_delim(file = "data\\ICGC数据\\exp_seq.all_projects.specimen.USonly.xena.tsv", delim = "\t", col_names = TRUE);
# 分组信息
cli <- read_delim(file = "data\\ICGC数据\\specimen.all_projects.tsv", delim = "\t", col_names = TRUE);
cli <- select(cli, c("specimen_type", "project_code"));
表达矩阵:
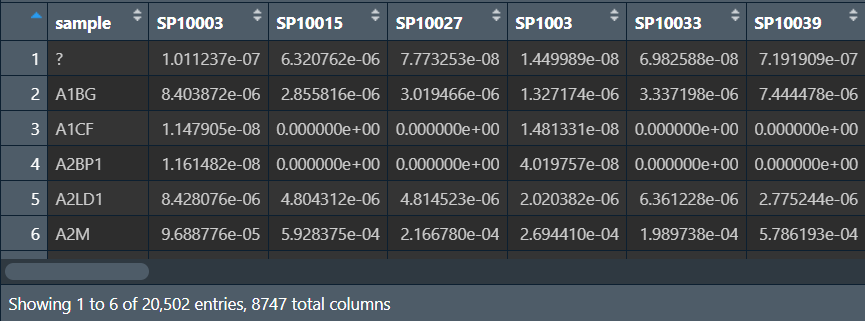
分组信息:
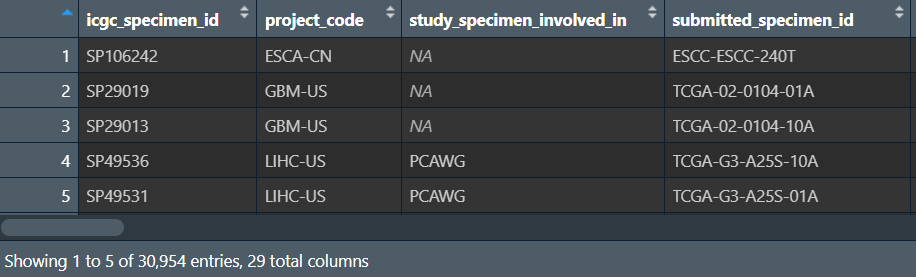
提取来自LUSC_CN、LUSC_KR和LUSC_US的样本:
LUSC_CN <- subset(cli, cli$project_code == c("LUSC-CN"));
LUSC_KR <- subset(cli, cli$project_code == c("LUSC-KR"));
LUSC_US <- subset(cli, cli$project_code == c("LUSC-US"));
# 合并
LUSC <- rbind(LUSC_CN, LUSC_KR, LUSC_US);
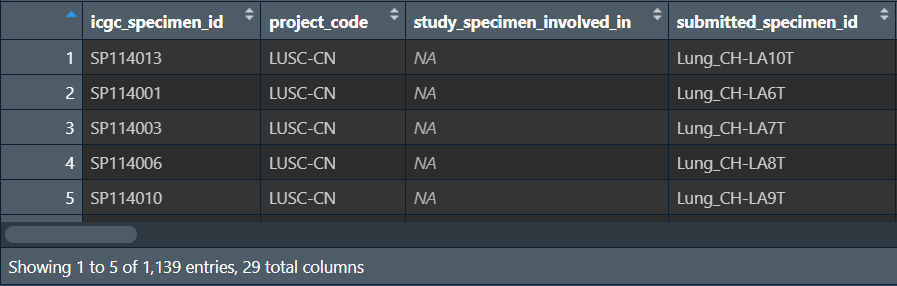
即筛选出LUSC(肺鳞癌)的样本,其它样本舍去
注:project_code中带US的(来自美国的)一般都是TCGA的样本,因此如果想结合TCGA分析,就不要使用这些带US的样本
获取正常和肿瘤样本,并取交集(LUSC的样本、正常/肿瘤的样本、表达矩阵中含有的样本):
注:正常样本的specimen_type列值为Normal - blood derived或Normal - tissue adjacent to primary,其它的都是肿瘤样本
# 正常/肿瘤组的分组信息
normal <- subset(cli, cli$specimen_type == c("Normal - blood derived", "Normal - tissue adjacent to primary"));
tumor <- subset(cli, cli$specimen_type != c("Normal - blood derived", "Normal - tissue adjacent to primary"));
# 正常/肿瘤组的样本名称
normalnames <- intersect(as.vector(normal[, 1])[["icgc_specimen_id"]], as.vector(LUSC[, 1])[["icgc_specimen_id"]]);
normalnames <- intersect(normalnames, colnames(exp));
tumornames <- intersect(as.vector(tumor[, 1])[["icgc_specimen_id"]], as.vector(LUSC[, 1])[["icgc_specimen_id"]]);
tumornames <- intersect(tumornames, colnames(exp));
# 正常/肿瘤组的表达矩阵
normalexp <- exp[, normalnames];
tumorexp <- exp[, tumornames];
tumorexp:
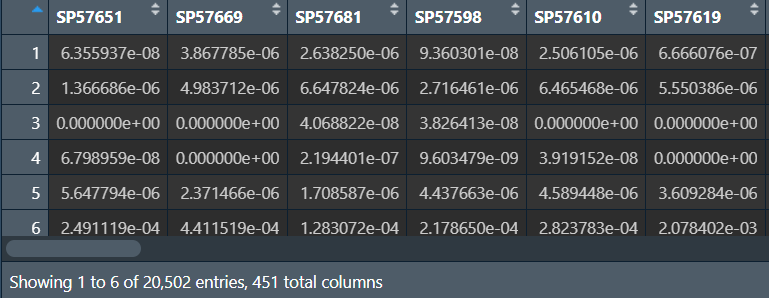
normalexp:

可以看到只剩下451和22个样本
合并正常/肿瘤组表达矩阵,同时加上基因名标记:
rt <- cbind(exp[, 1, drop = F], normalexp, tumorexp);
rt <- rt[-1, ]; # 删除第一行(基因名为?的那一行,是无用信息)
write.table(rt, 'data\\ICGC数据\\matrix.txt', sep = "\t", quote = F, row.names = F);
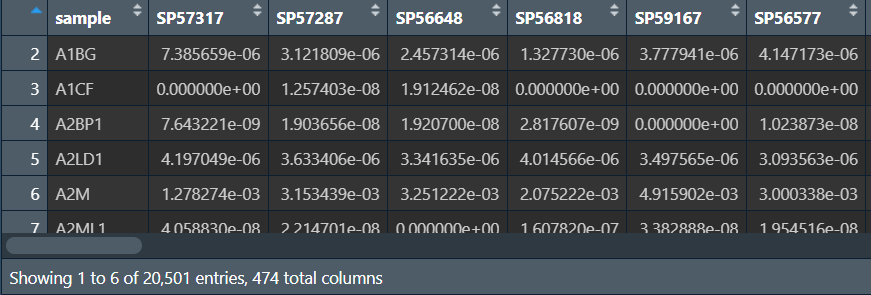
总共22+451+1=474列(正常+肿瘤+基因名),列名是样本名,行名(第一列)是基因名,数据是各基因在各样本中的表达量
临床数据
library(tidyverse);
读取临床信息和表达矩阵,合并:
# 临床信息
cli <- read_delim(file = "data\\ICGC数据\\donor.all_projects.overallSurvival_transfer_specimen", delim = "\t", col_names = TRUE);
cli <- as.data.frame(cli);
rownames(cli) <- cli[, 1]; # 行名为样本名
# 表达矩阵
data <- read.table("data\\ICGC数据\\matrix.txt", header = T, sep = "\t", check.names = F, row.names = 1);
# 合并
samesample <- intersect(rownames(cli), colnames(data));
cli.samesample <- cli[samesample, ];

去除NA值,仅保留生存状态和生存时间列:
cli.samesample <- na.omit(cli.samesample);
cli.samesample <- cli.samesample[, c(3, 4)];
# 保存
write.table(
data.frame(
ID = rownames(cli.samesample),
cli.samesample
),
file = "data\\ICGC数据\\clinical.time.txt",
sep = "\t", quote = F, row.names = F, col.names = T
);
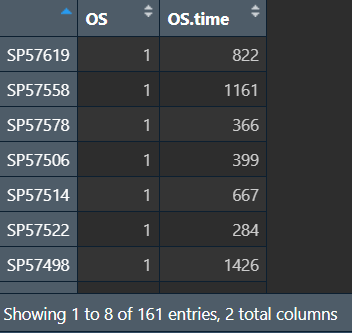
自定义通路富集打分
需要数据:tpm表达矩阵,自定义基因集(第一列是基因集名称,第二列是对基因集的描述,如果没有就设为NA,后面是各基因集包含的基因)
library(limma);
library(GSEABase);
library(GSVA);
library(pheatmap);
library(reshape2);
library(ggpubr);
library(readxl);
读取表达矩阵和基因集:
# 表达矩阵
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", header = T, sep = "\t", check.names = F,row.names = 1);
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
# 基因集
geneSets_xlsx <- read_excel("data\\genesets.xlsx", col_names = F);
# 转为gmt文件,并读取
write.table(geneSets_xlsx, file = "save_data\\my_genesets.gmt", sep = "\t", row.names = F, col.names = F, quote = F);
geneSets <- getGmt("save_data\\my_genesets.gmt", geneIdType = SymbolIdentifier());
打分并标准化:
# 打分
gsvapar <- gsvaParam(data, geneSets, kcdf = 'Gaussian', absRanking = TRUE);
gsvaResult <- gsva(gsvapar);
# 标准化
normalize <- function(x){return((x-min(x))/(max(x)-min(x)));}
gsvaResult <- normalize(gsvaResult);
# 保存
gsvaOut <- rbind(id = colnames(gsvaResult), gsvaResult);
write.table(gsvaOut, file = "save_data\\ssgseaOut2.txt", sep = "\t", quote = F, col.names = F);
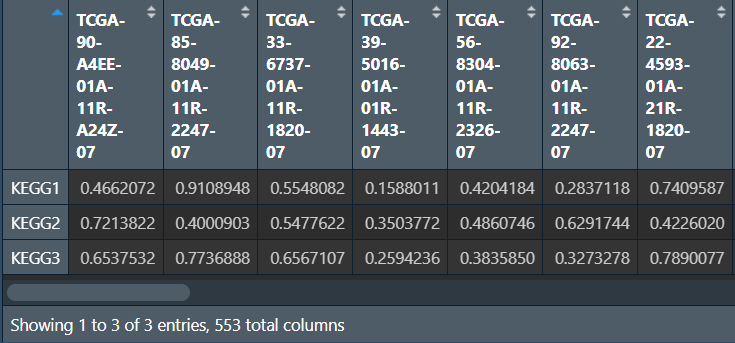
这是不同的样本在三个自定义通路中的得分情况
分组,并提取差异显著的通路:
# 分组
group <- sapply(strsplit(colnames(data), '\\-'), "[", 4);
group <- sapply(strsplit(group, ''), "[", 1);
group <- gsub("2", "1", group);
conNum <- length(group[group==1]);
treatNum <- length(group[group==0]);
Type <- c(rep(1, conNum), rep(2, treatNum));
# 根据正常和肿瘤排序
gsvaResult1 <- gsvaResult[, group == 1];
gsvaResult2 <- gsvaResult[, group == 0];
gsvaResult <- cbind(gsvaResult1, gsvaResult2);
gsvaResult <- cbind(t(gsvaResult), Type);
# 获取差异显著的通路
sigGene <- c();
for(i in colnames(gsvaResult)[1:(ncol(gsvaResult)-1)]){
test <- wilcox.test(gsvaResult[,i] ~ gsvaResult[,"Type"]);
pvalue <- test$p.value;
if(pvalue<0.05){ # p值阈值,可修改
sigGene <- c(sigGene, i);
}
}
# 提取差异显著的通路
hmExp <- gsvaResult[, sigGene];
Type <- c(rep("Normal", conNum), rep("Tumor", treatNum));
names(Type) <- rownames(gsvaResult);
Type <- as.data.frame(Type);
hmExp <- t(hmExp);
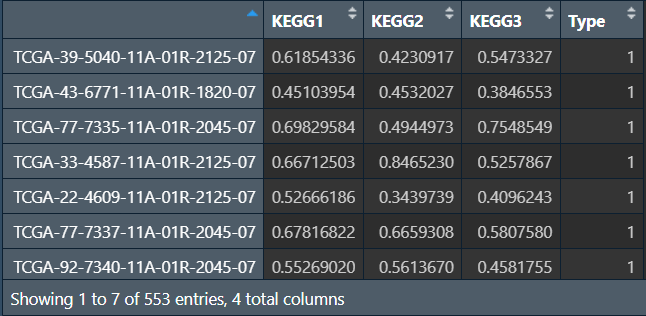
画箱线图:
# 输入数据格式转换(宽变长)
hmExp2 <- t(hmExp);
hmExp2 <- cbind(hmExp2, Type);
rt <- melt(hmExp2, id.vars = c("Type"));
colnames(rt) <- c("Type", "Genesets", "Expression");
# 设置比较组
group <- levels(factor(rt$Type));
rt$Type <- factor(rt$Type, levels = c("Normal", "Tumor"));
# 画图
boxplot <- ggboxplot(
rt, x = "Genesets", y = "Expression",
fill = "Type",
xlab = "", ylab = "Score",
legend.title = "Type",
width = 0.8,
palette = c("DodgerBlue1", "Firebrick2") # 修改颜色
) +
rotate_x_text(50) +
stat_compare_means(
aes(group = Type),
method = "wilcox.test",
symnum.args = list(
cutpoints = c(0, 0.001, 0.01, 0.05, 1),
symbols = c("***", "**", "*", "ns")
),
label = "p.signif"
) +
theme(
axis.text = element_text(face = "bold.italic", colour = "#441718", size = 16),
axis.title = element_text(face = "bold.italic", colour = "#441718",size = 16),
axis.line = element_blank(),
plot.title = element_text(face = "bold.italic",colour = "#441718",size = 16),
legend.text = element_text(face = "bold.italic"),
panel.border = element_rect(fill = NA, color = "#35A79D", size = 1.5, linetype = "solid"),
panel.background = element_rect(fill = "#F1F6FC"),
panel.grid.major = element_line(color = "#CFD3D6", size = .5, linetype = "dotdash"),
legend.title = element_text(face = "bold.italic", size = 13)
);
# 输出图片
pdf(file = "save_data\\ggboxplot.pdf", width = 8, height = 6);
print(boxplot);
dev.off();
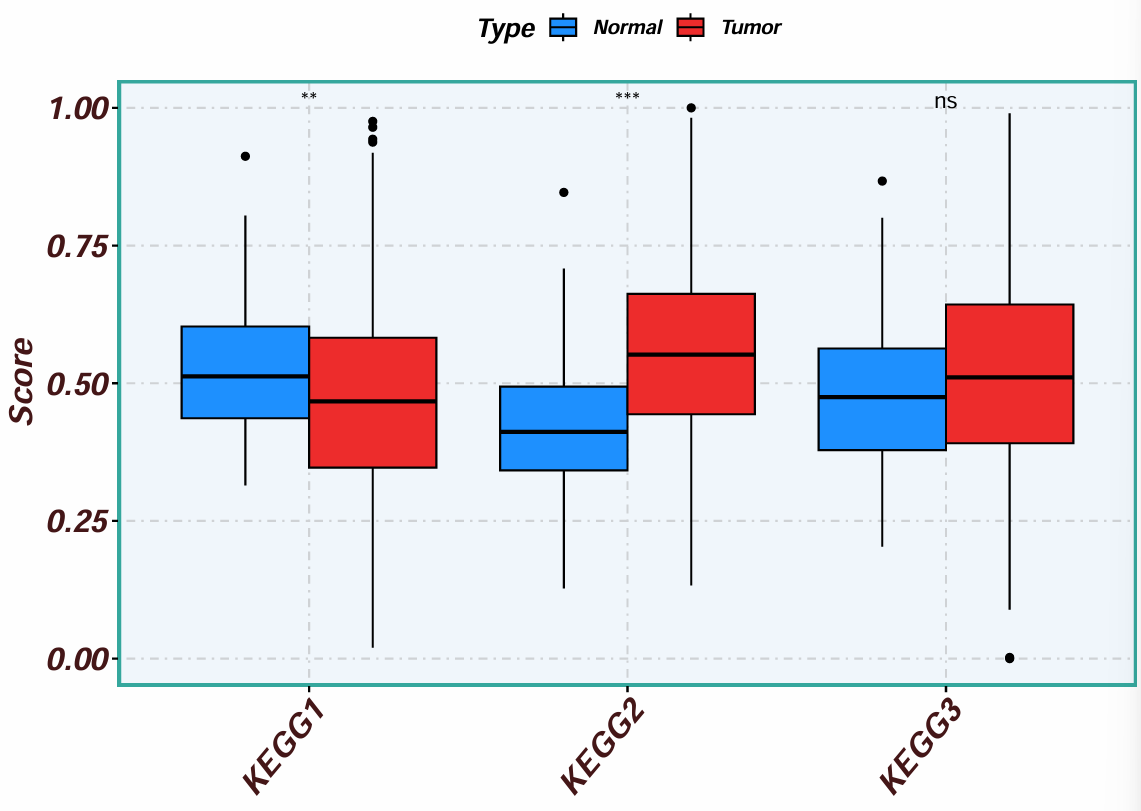
横坐标是三个自定义通路,纵坐标是打分分值(富集情况),分组是正常/肿瘤组
IPS预测免疫治疗反应
免疫表型评分(Immunophenoscore, IPS)可以确定肿瘤的免疫原性,并预测多种类型的肿瘤对免疫检查点抑制剂治疗的反应
TCGA
进入tcia官网,选择肿瘤类型,以LUSC为例,点击柱状图中LUSC的那个柱子
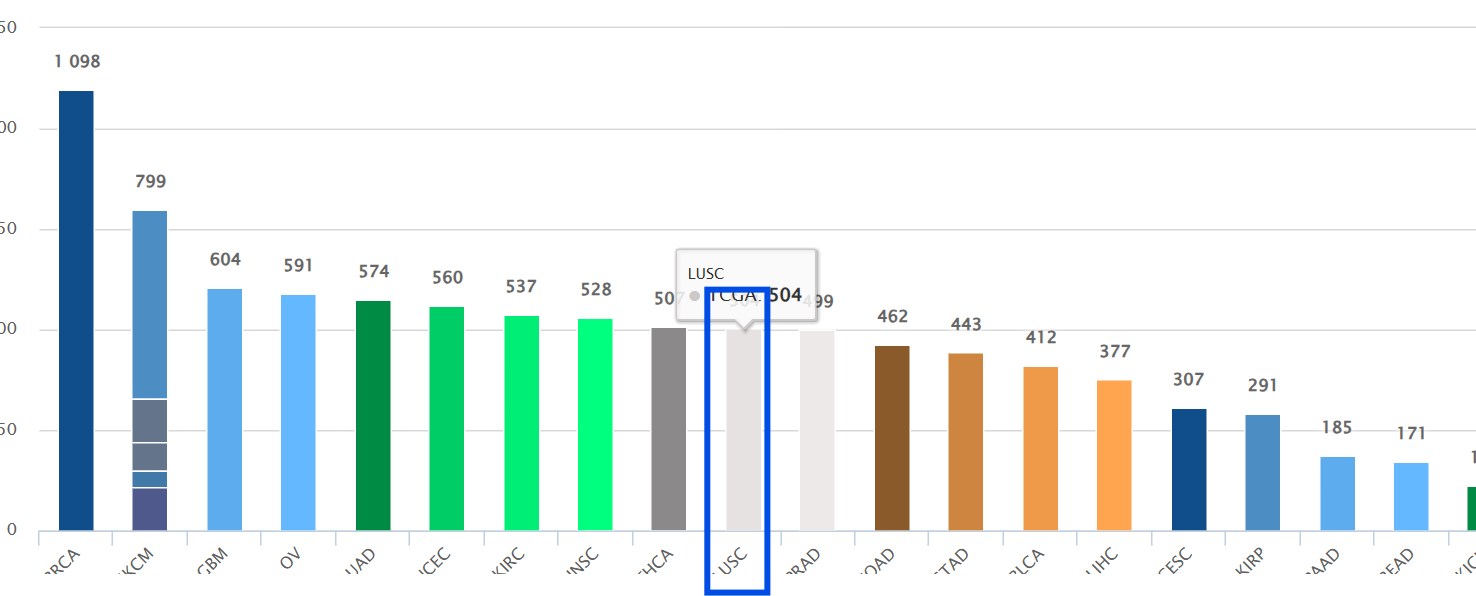
点击Export all to TSV下载
除此之外,还需要tpm表达矩阵
library(reshape2);
library(ggpubr);
读取临床数据和表达矩阵,合并:
# 临床数据
tcia <- read.table("data\\TCIA-ClinicalData.tsv", header = T, sep = "\t", check.names = F, row.names = 1);
tcia <- tcia[, c("ips_ctla4_neg_pd1_neg", "ips_ctla4_neg_pd1_pos", "ips_ctla4_pos_pd1_neg", "ips_ctla4_pos_pd1_pos")]; # 仅选取这4列与免疫抑制剂相关的
# 表达矩阵
data <- read.table("save_data\\TCGA_LUSC_TPM.txt", header = T, sep = "\t", check.names = F, row.names = 1);
# 仅保留肿瘤样本
group <- sapply(strsplit(colnames(data), "\\-"), "[", 4);
group <- sapply(strsplit(group,""), "[", 1);
data <- data[, group == 0];
# 改样本名
colnames(data) <- gsub("(.*?)\\-(.*?)\\-(.*?)\\-(.*)", "\\1\\-\\2\\-\\3", colnames(data));
# 转置
data <- t(data);
# 合并
sameSample <- intersect(row.names(tcia), row.names(data));
tcia <- tcia[sameSample, , drop=F];
# 以CTLA4表达量为分组依据
data <- data[sameSample, "CTLA4", drop=F];
data <- cbind(tcia,data);
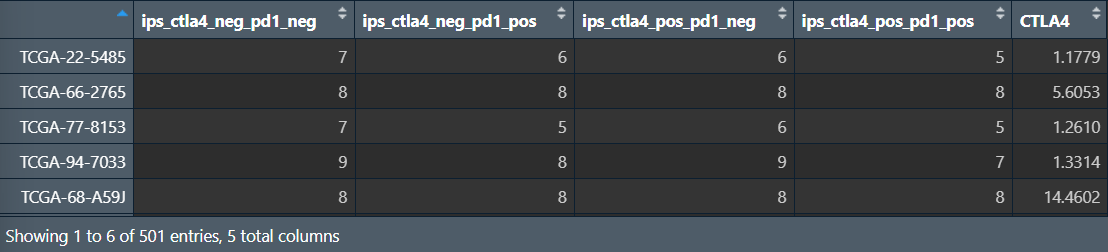
按CTLA4表达量分组,并转换数据格式(宽变长):
# 分组
data$group <- ifelse(
data[, "CTLA4"]>quantile(data[, "CTLA4"], seq(0, 1, 1/2))[2],
"high", "low"
);
data <- data[, -5]; # 去掉基因表达量那列(只留下是高/低表达组标识即可)
# 宽变长
rt <- melt(data, id.vars = c("group"));
colnames(rt) <- c("group", "Genesets", "Expression");
group <- levels(factor(rt$group));
rt$group <- factor(rt$group, levels = group);
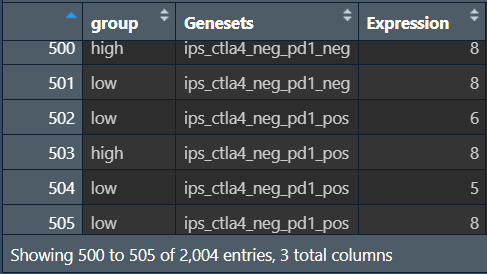
画提琴图:
violin <- ggviolin(
rt, x = "Genesets", y = "Expression",
fill = "group",
xlab = "", ylab = "Score",
legend.title = "group",
# width = 0.8,
palette = c("DodgerBlue1", "Firebrick2")
) +
rotate_x_text(50) +
stat_compare_means(
aes(group = group),
method = "wilcox.test",
symnum.args = list(
cutpoints = c(0, 0.001, 0.01, 0.05, 1),
symbols = c("***", "**", "*", "ns")
),
label = "p.signif"
) +
theme(
axis.text = element_text(face = "bold.italic", colour = "#441718", size = 16),
axis.title = element_text(face = "bold.italic", colour = "#441718", size = 16),
axis.line = element_blank(),
plot.title = element_text(face = "bold.italic", colour = "#441718", size = 16),
legend.text = element_text(face = "bold.italic"),
panel.border = element_rect(fill = NA, color = "#35A79D", size = 1.5, linetype = "solid"),
panel.background = element_rect(fill = "#F1F6FC"),
panel.grid.major = element_line(color = "#CFD3D6", size = .5, linetype = "dotdash"),
legend.title = element_text(face = "bold.italic", size = 13)
);
# 输出
pdf(file = "save_data\\ggviolin_ips.pdf", width = 8, height = 7);
print(violin);
dev.off();
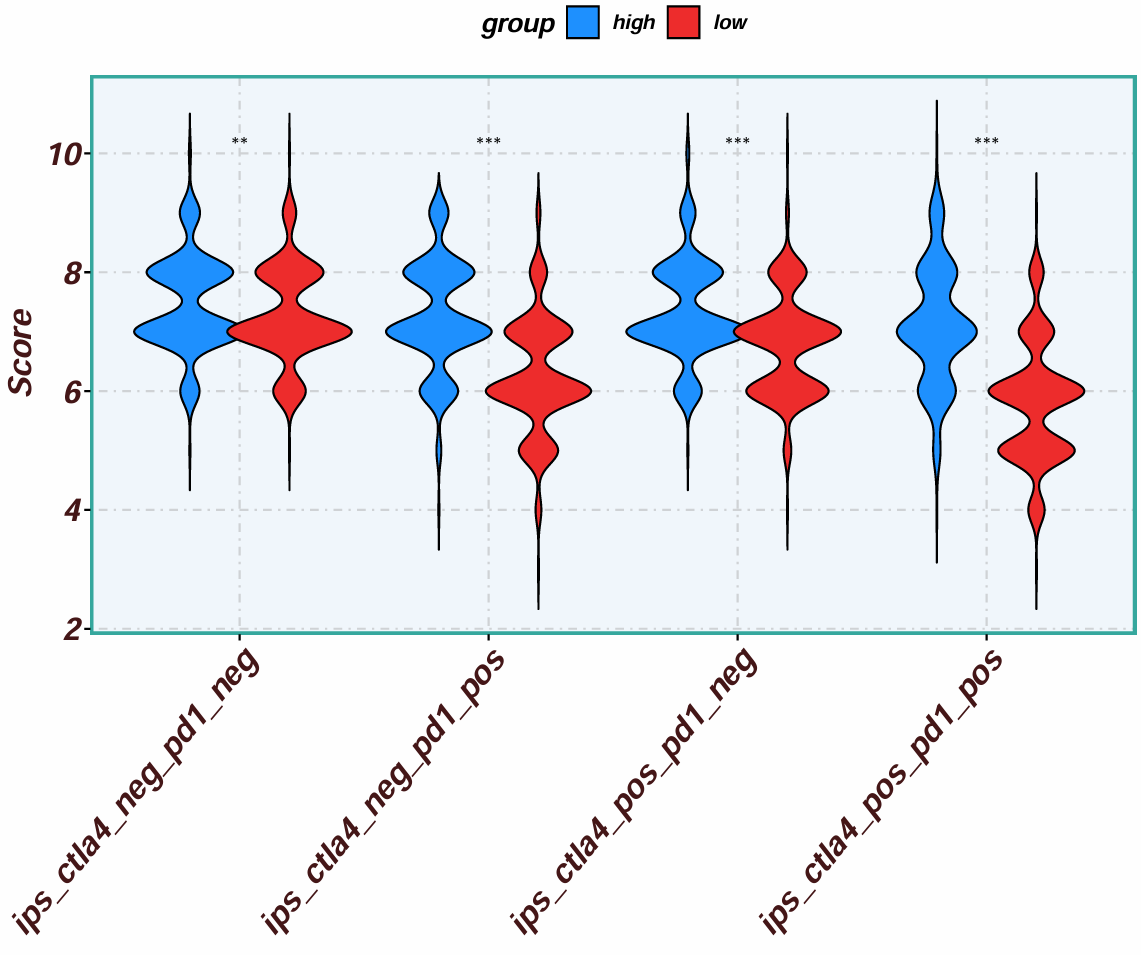
按CTLA4表达量高低分组;横坐标标识了使用了哪种免疫抑制剂,pos是使用、neg是没使用,比如ctla4_pos_pd1_pos就是使用了ctla4和pd1这两种免疫抑制剂;纵坐标是得分,得分越高,则该组对指定的抑制剂就越敏感
非TCGA
需要数据:GEO数据库的GSE74777数据集
if(!require("IOBR", quietly = T))
{
library("devtools");
devtools::install_github("spatstat/spatstat.sparse");
devtools::install_github("IOBR/IOBR");
}
library(IOBR);
library(tidyverse);
library(ggpubr);
library(limma);
library(reshape2);
读入表达矩阵,并进行ips分析:
# 表达矩阵
data <- read.table("data\\GSE74777\\GSE74777.txt", header = T, sep = "\t", check.names = F, row.names = 1);
# 标准化
data <- normalizeBetweenArrays(data)
# ips分析
ips <- deconvo_tme(eset = data, method = "ips", plot = FALSE);
# 转置
data <- t(data);
# 合并
data <- cbind(ips, data[, "CTLA4", drop=F]);
注:deconvo_tme还可以做其它很多的免疫分析
按CTLA4表达量分组,并转换数据格式(宽变长):与上面TCGA的类似
# 分组
data$group <- ifelse(
data[,"CTLA4"]>quantile(data[, "CTLA4"], seq(0, 1, 1/2))[2],
"high", "low"
);
data <- data[, -c(1, 8)];
# 宽变长
rt <- melt(data, id.vars = c("group"));
colnames(rt) <- c("group", "Genesets", "Expression");
group <- levels(factor(rt$group));
rt$group <- factor(rt$group, levels = group);
画箱线图:(与预测药物敏感性的代码相同)
boxplot <- ggboxplot(
rt, x = "Genesets", y = "Expression",
fill = "group",
xlab = "", ylab = "Score",
legend.title = "group",
width = 0.8,
palette = c("DodgerBlue1","Firebrick2")
) +
rotate_x_text(50) +
stat_compare_means(
aes(group = group),
method = "wilcox.test",
symnum.args = list(
cutpoints = c(0, 0.001, 0.01, 0.05, 1),
symbols = c("***", "**", "*", "ns")
),
label = "p.signif"
) +
theme(
axis.text = element_text(
face = "bold.italic",
colour = "#441718",
size = 16
),
axis.title = element_text(
face = "bold.italic",
colour = "#441718",
size = 16
),
axis.line = element_blank(),
plot.title = element_text(
face = "bold.italic",
colour = "#441718",
size = 16
),
legend.text = element_text(face = "bold.italic"),
panel.border = element_rect(
fill = NA,
color = "#35A79D",
size = 1.5,
linetype = "solid"
),
panel.background = element_rect(fill = "#F1F6FC"),
panel.grid.major = element_line(
color = "#CFD3D6",
size = .5,
linetype = "dotdash"
),
legend.title = element_text(
face = "bold.italic",
size = 13
)
);
#输出
pdf(file = "save_data\\ggboxplot_ips.pdf", width = 8, height = 6);
print(boxplot);
dev.off();
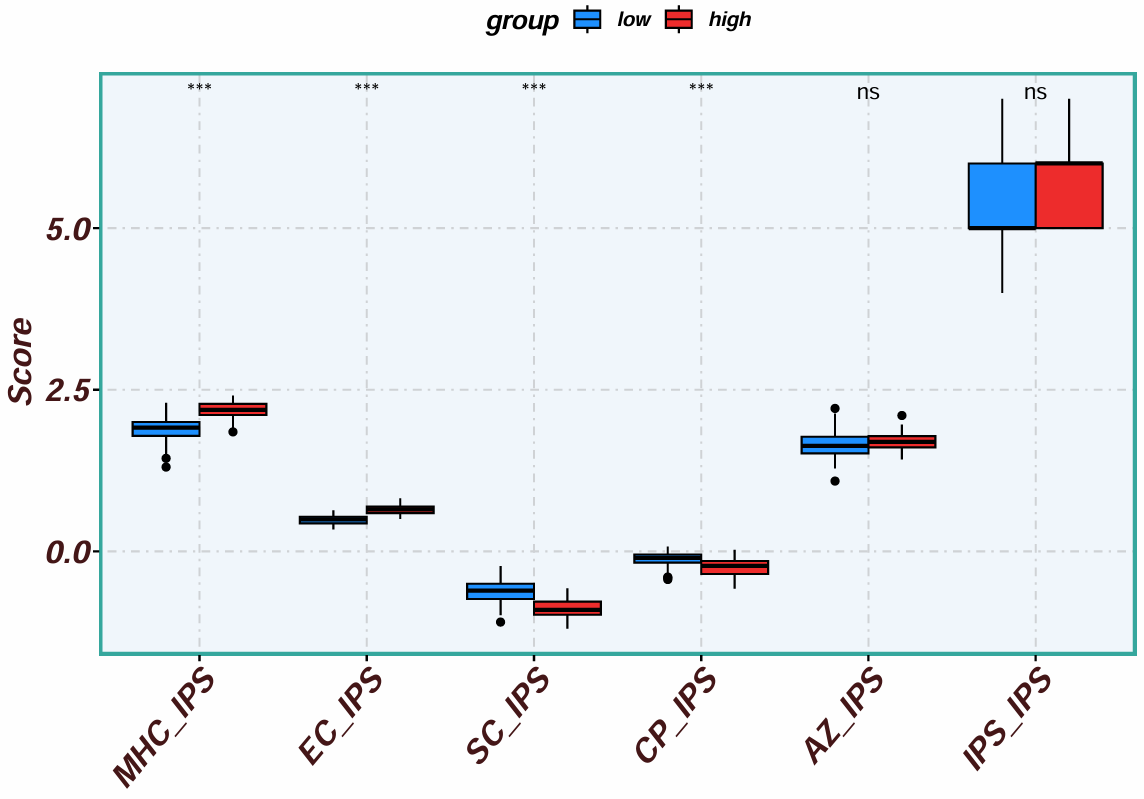
根据CTLA4基因的表达高/低分组,纵坐标是评分,横坐标是不同的免疫表型,有4个主要的参数:
-
MHC分子(抗原呈递)(MHC molecules, MHC)
-
效应细胞(Effector cells, EC)
-
抑制性细胞(Suppressor cells, SC)
-
免疫检查点(immune checkpoints, CP) / 免疫调节分子(Immunomodulators, CP)