b站生信课程02-6
r-bioinfolessonb站生信课程TCGA及GEO数据挖掘入门必看P44-P55笔记——基因在染色体位置、拷贝数突变频率、二分类列线图、miRNA下游靶基因、二分类cox分析、生存状态详细分析、TCGA和GEO数据下载补充
写在前面:本篇教程来自b站课程TCGA及GEO数据挖掘入门必看 P44-P55 相关资料下载
基因所在染色体位置
需要数据:一个标识各基因在染色体上位置的文件Ref.txt,要标识的基因名称(这里以多因素cox回归得到的基因为例)
if(!require("RCircos", quietly = T))
{
library(BiocManager);
BiocManager::install("RCircos");
}
library("RCircos");
读取数据:基因位置信息、基因名称
data(UCSC.HG19.Human.CytoBandIdeogram); # 内置的人类染色体数据
data <- read.table("save_data\\multiCox.txt", check.names = F, row.names = 1, sep = '\t', header = T); # 基因名称
dataref <- read.table("data\\Ref.txt", header = T, sep = "\t", check.names = F, row.names = 1); # 基因位置信息
# 合并
samegene <- intersect(rownames(dataref), rownames(data));
generef <- dataref[samegene, ];
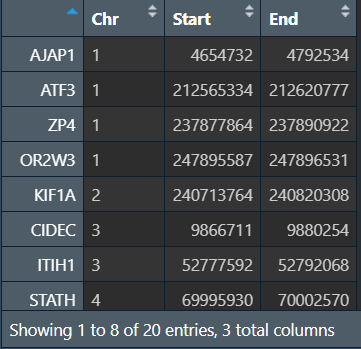
修改格式:将行名(基因名)提取为Gene列
generef2 <- cbind(generef[, 1:3], rownames(generef));
colnames(generef2) <- c("Chromosome", "chromStart", "chromEnd", "Gene"); # 改列名
generef2[, 1] <- paste("chr", generef2[, 1], sep = ""); # 在染色体名称前加"chr"
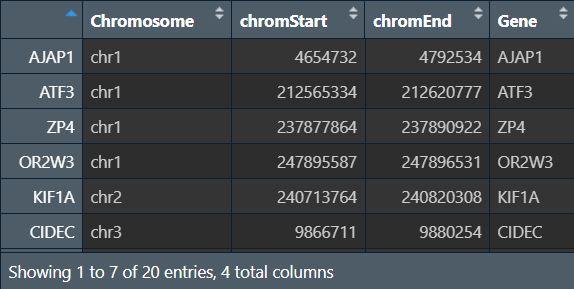
画图:
pdf(file = "save_data\\Circle.pdf", width = 7, height = 7);
cyto.info <- UCSC.HG19.Human.CytoBandIdeogram;
chr.exclude <- NULL;
tracks.inside <- 4;
tracks.outside <- 0;
RCircos.Set.Core.Components(cyto.info, chr.exclude, tracks.inside, tracks.outside);
chr.exclude <- NULL;
RCircos.Set.Plot.Area();
RCircos.Chromosome.Ideogram.Plot();
side <- "in";
track.num <- 1;
RCircos.Gene.Connector.Plot(generef2, track.num, side);
name.col <- 4;
track.num <- 2;
RCircos.Gene.Name.Plot(generef2, name.col,track.num, side);
dev.off();
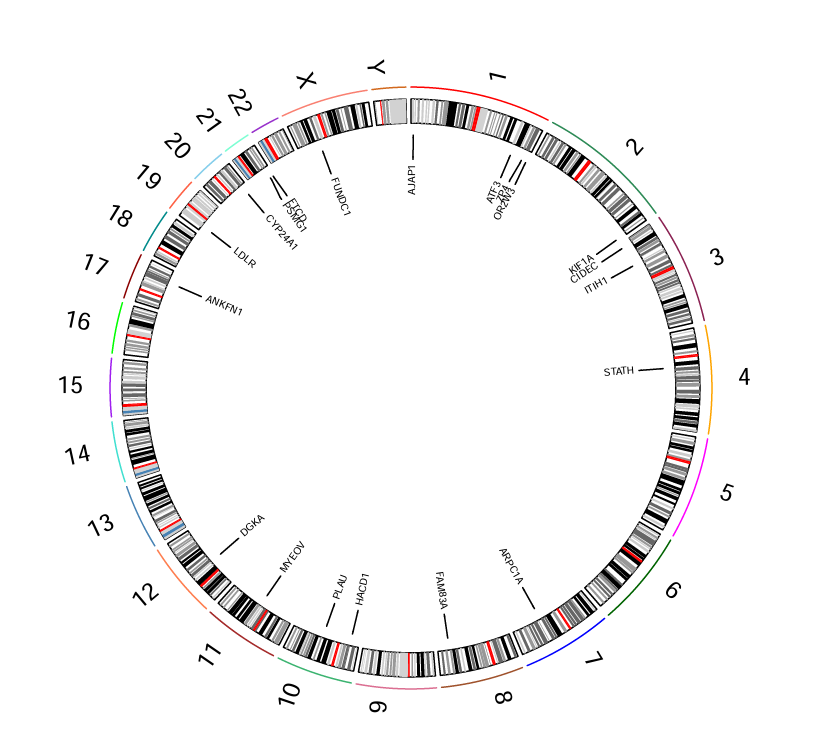
外圈代表23对染色体,内圈代表不同基因在染色体上的位置
拷贝数突变频率
进入ucsc官网,找到GDC TCGA Lung Squamous Cell Carcinoma (LUSC)
下载GISTIC - focal score by gene (n=503) GDC Hub
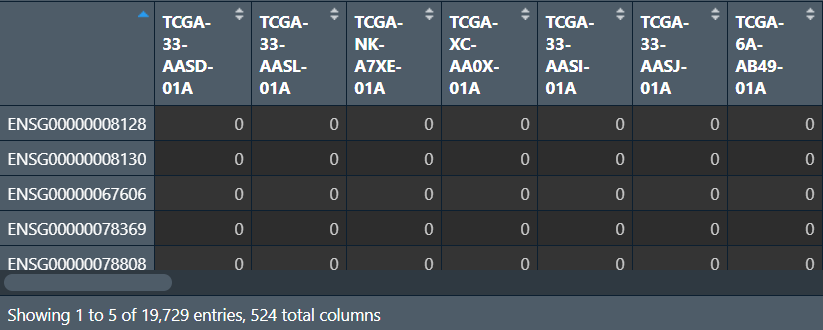
下载后解压,行名是基因symbol,列名是样本名
需要数据:刚才下载的突变数据、基因列表(这里以多因素cox回归得到的基因为例)、基因组注释文件GRCh38.gtf(下载方法)
library(maftools);
library(rtracklayer);
library(stringr);
读取突变数据:
CNV <- read.table("data\\TCGA-LUSC.gistic.tsv", header = T, sep = "\t", check.names = F, row.names = 1);
# 更改行名,删除.之后的数
rownames(CNV) <- unlist(lapply(rownames(CNV), function(x){
str_split(string = x, pattern = "\\.")[[1]][1];
}));
读取注释信息,并将基因id转为基因名:(类似于GEO数据的注释)
gtf <- rtracklayer::import('data\\GRCh38.gtf');
gtf <- as.data.frame(gtf);
ids <- gtf[, c("gene_id", "gene_name")];
colnames(ids) <- c('probe_id', 'symbol'); # 更改列名
ids <- aggregate(. ~ probe_id, data = ids, max); # 删除重复id
ids <- ids[ids$probe_id %in% rownames(CNV), ]; # 取出ids在CNV行名中的基因id
CNV <- CNV[ids$probe_id, ]; # 根据行名(基因id)取出CNV在ids探针id中的行
CNV <- cbind(ids, CNV); # 合并
CNV <- aggregate(. ~ symbol, data = CNV, FUN = max); # 去重
rownames(CNV) <- CNV[, 1]; # 改行名为基因名
CNV <- CNV[, -c(1, 2)]; # 去除前两列(基因id和基因名)
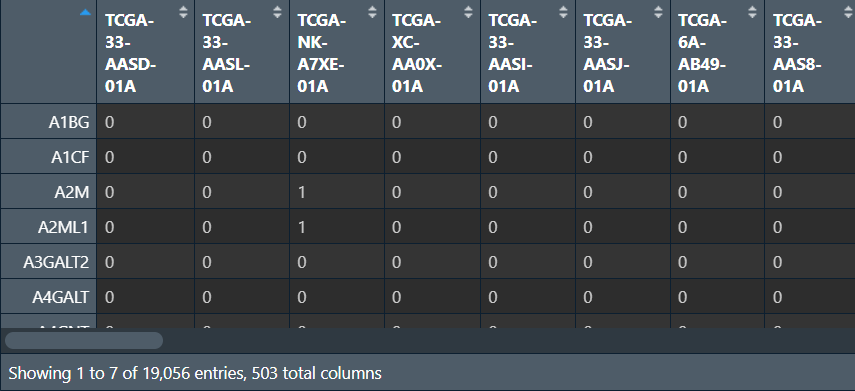
读取基因名称,合并,并计算GAIN和LOSS值:
data <- read.table("save_data\\multiCox.txt", check.names = F, row.names = 1, sep = '\t', header = T); # 基因名称
samegenes <- intersect(rownames(data),rownames(CNV)); # 共有基因
rt <- CNV[samegenes, ]; # 提取
# 计算GAIN和LOSS值
GAIN <- rowSums(rt>0);
LOSS <- rowSums(rt<0);
GAIN <- GAIN/ncol(rt)*100;
LOSS <- LOSS/ncol(rt)*100;
data <- cbind(GAIN, LOSS);
# 按GAIN值从大到小排序
data <- data[order(data[, "GAIN"], decreasing = T), ];
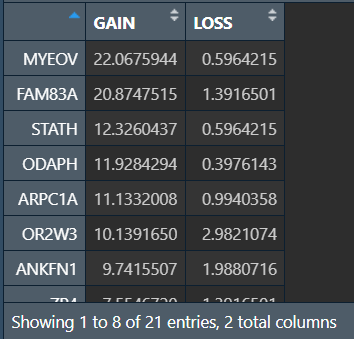
行名是基因名,列分别是GAIN和LOSS值
画图:
data.max <- apply(data, 1, max); # 每个基因GAIN和LOSS两者中的的最大值
pdf(file = "save_data\\CNV.frequency.pdf", width = 9, height = 6);
cex <- 1.3;
par(
cex.lab = cex,
cex.axis = cex,
font.axis = 2,
las = 1,
xpd = T
);
bar <- barplot(
data.max,
col = "grey80",
border = NA,
xlab = "", ylab = "CNV.frequency(%)",
space = 1.5,
xaxt = "n",
ylim = c(0, 1.2*max(data.max))
);
points(bar, data[, "GAIN"], pch = 20, col = 2, cex = 3);
points(bar, data[, "LOSS"], pch = 20, col = 3, cex = 3);
legend(
"top",
legend = c('GAIN','LOSS'),
col = 2:3,
pch = 20,
bty = "n",
cex = 2,
ncol = 2
);
par(srt = 45);
text(bar, par('usr')[3]-0.2, rownames(data), adj = 1, cex = 0.8);
dev.off();
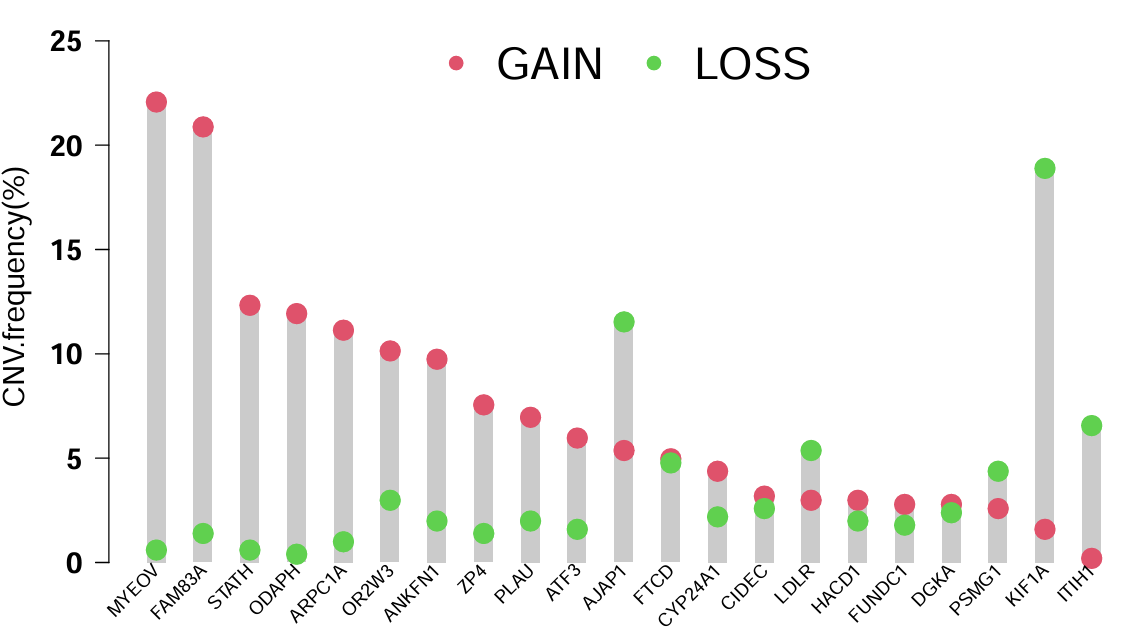
横坐标是基因,纵坐标是拷贝数突变频率,gain和loss分别代表增加和丢失
TCGA甲基化数据下载和整理
library(stringr);
library(tidyverse);
读取json文件,获取样本名称和文件名称的对照:
json <- jsonlite::fromJSON("data\\甲基化数据\\metadata.cart.2023-12-31.json");
# 样本名称
sample_id <- sapply(json$associated_entities, function(x){x[, 1]});
# 对应的txt文件名称
file_name <- json$file_name;
# 合并成df
file_sample <- data.frame(sample_id, file_name);
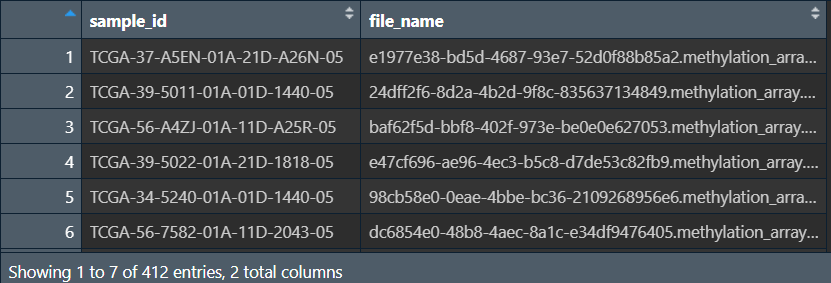
获取每个txt的文件名称:
count_file <- list.files('data\\甲基化数据\\gdc_download_20231230_164551.556154\\', pattern = '*level3betas.txt', recursive = TRUE);
count_file_name <- strsplit(count_file, split='/');
count_file_name <- sapply(count_file_name, function(x){x[2]});
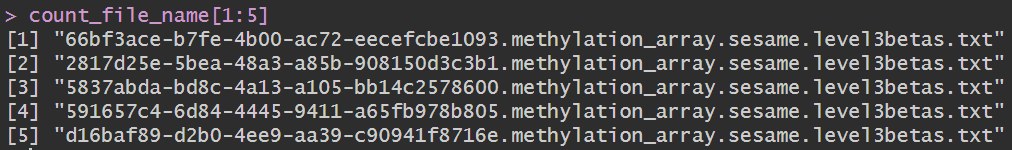
读取数据:结果矩阵中列名是样本名,行名是甲基化位点
# 先读取一个文件,看看有多少行
test_data <- read.delim(
paste0('data\\甲基化数据\\gdc_download_20231230_164551.556154\\', count_file[1]),
fill = TRUE, header = FALSE, row.names = 1
);
# 结果矩阵
matrix <- data.frame(matrix(nrow = nrow(test_data), ncol = 0));
# 逐个读取及合并
for (i in 1:length(count_file)){
# 根据文件名读取数据
data <- read.delim(
paste0('data\\甲基化数据\\gdc_download_20231230_164551.556154\\', count_file[i]),
fill = TRUE, header = FALSE, row.names = 1
);
# 找该文件名对应的样本名
index <- which(file_sample$file_name==count_file_name[i]); # 对应的样本名的索引
sample_name <- file_sample$sample_id[index]; # 对应的样本名
colnames(data) <- sample_name; # 作列名
matrix <- cbind(matrix, data); # 添加到结果矩阵中
}
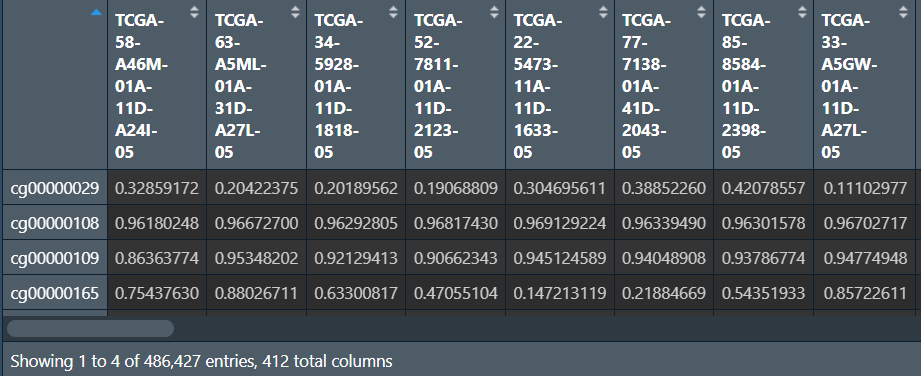
删除NA并导出:
matrix <- na.omit(matrix); # 删除NA
save_df <- data.frame(ID = rownames(matrix), matrix);
colnames(save_df) <- gsub('[.]', '-', colnames(save_df));
# 保存
write.table(save_df, 'data\\甲基化数据\\methylation.450.txt', sep = "\t", quote = F, row.names = F);
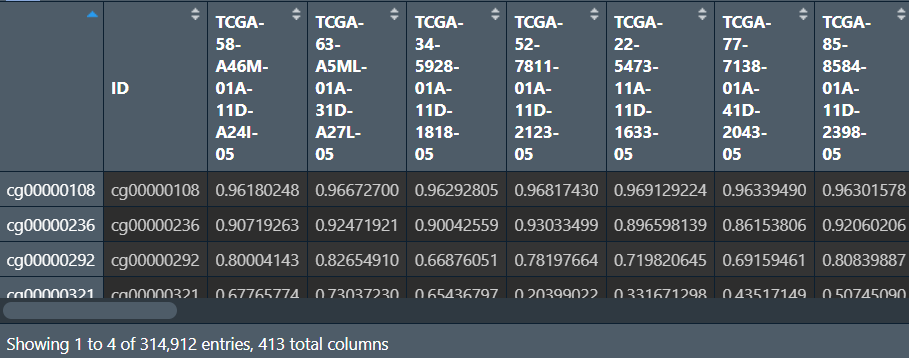
行名(id列)是探针id,列名是样本名,数据值从0到1代表甲基化程度增加(1为完全甲基化,0是完全没有甲基化
补充:也可以从UCSC中下载:
找到TCGA Lung Squamous Cell Carcinoma (LUSC)


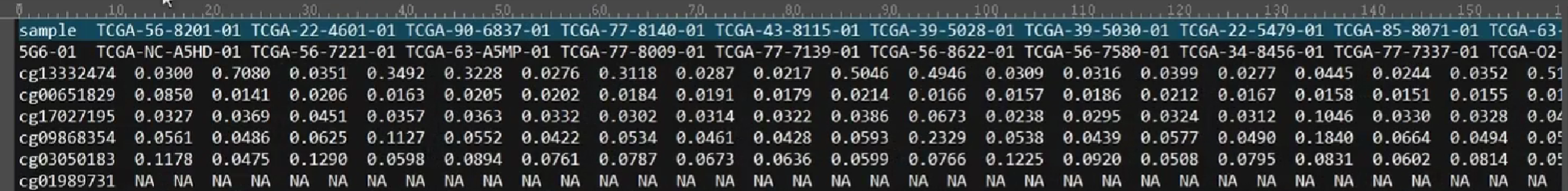
下载的文件与刚才整理的相同,都是行名是样本名,列名是甲基化编号,不过未删除NA值
logistic列线图
之前的是使用cox方法(有多个因素,预测模型),对于单一的二分类的变量(比如正常/肿瘤组、控制/治疗组,诊断模型),可以使用logistic方法
需要数据:GSE30219.txt表达矩阵(行名为基因名,列名为样本名,数据为表达量),之前在GEO差异表达分析中得到的Control.txt和Treat.txt分组(控制/治疗组),当然也可以使用正常/肿瘤组
if(!require("rmda", quietly = T))
{
install.packages("rmda");
}
library(rms);
library("rmda");
library(limma);
读取表达矩阵并标准化:
data <- read.table("data\\GSE30219\\GSE30219.txt", header = T, sep = "\t", check.names = F, row.names = 1);
# 转化为matrix
dimnames <- list(rownames(data), colnames(data));
data <- matrix(as.numeric(as.matrix(data)), nrow = nrow(data), dimnames = dimnames);
# 去除低表达的基因
data <- data[rowMeans(data)>1, ];
# 标准化
data <- normalizeBetweenArrays(data);
# 保存
write.table(
data.frame(ID = rownames(data), data),
file = "data\\GSE30219\\normalize.txt",
sep = "\t", quote = F, row.names = F
);
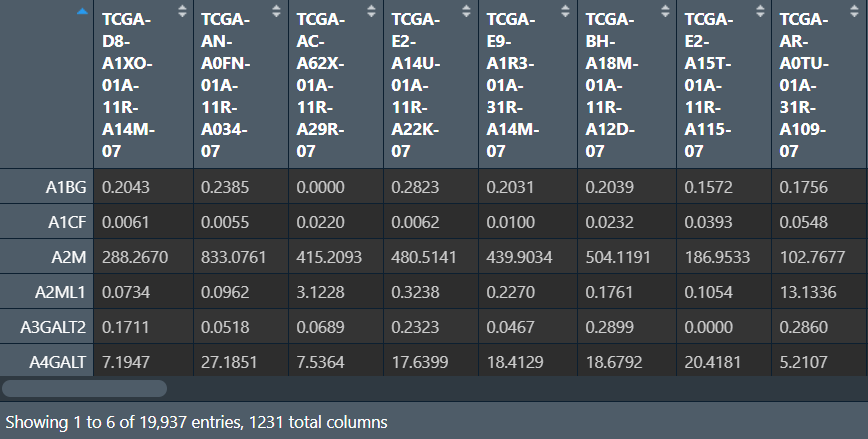
选取基因,读取分组信息:
gene <- c("A1BG","A1CF","A2M","A2ML1","A2MP1","A4GALT"); # 基因列表
#获取基因
data <- data[gene, ]; # 提取表达矩阵
data <- t(data);
# 读取分组信息
Control <- read.table("data\\GSE30219\\Control.txt", header = F, sep = "\t", check.names = F);
Treat <- read.table("data\\GSE30219\\Treat.txt", header = F, sep = "\t", check.names = F);
# 按照control和treat排序
data <- data[c(Control[, 1], Treat[, 1]), ];
# 设置分组信息
conNum <- length(rownames(Control));
treatNum <- length(rownames(Treat));
group <- c(rep("Control", conNum), rep("Treat", treatNum));
# 合并
rt <- cbind(as.data.frame(data), Type = group);
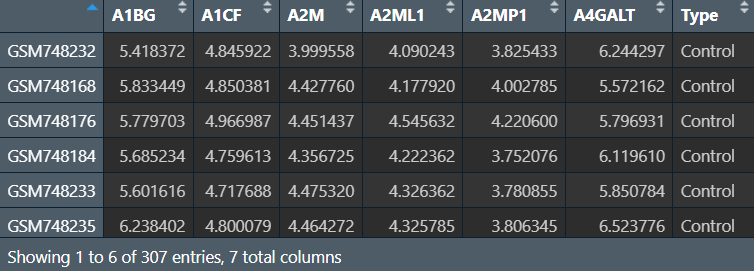
前6列是基因名,最后一列是分组信息,行名是样本名
构建模型:
model <- paste0("Type", "~", paste(colnames(data), collapse="+")); # 模型公式:Type~A1BG+A1CF+A2M+A2ML1+A2MP1+A4GALT
ddist <- datadist(rt);
options(datadist = "ddist"); # 为模型设置数据
lrmModel <- lrm(as.formula(model), data = rt, x = T, y = T);
nomo <- nomogram(
lrmModel,
fun = plogis,
fun.at = c(0.1, 0.5, 0.8, 0.99), # 展示的概率(x轴值)
lp = F,
funlabel = "Risk of Disease"
);
列线图:
pdf("save_data\\Nom.pdf", width = 9, height = 6);
plot(nomo);
dev.off();

最上面的points是得分,中间的6行是随机挑选的6个基因,横坐标对应着不同的得分。以A1BG这行为例,它横坐标为5时对应的points为0、为8.5时对应的points为100,即表达量为5时得分为0、表达量为8.5时得分为100。total points是总得分,最下面的risk of disease是患病几率,如果总得分为0,它对应着的患病几率为0,就是不会患病;如果总得分为60,它对应着的患病几率为0.5,就是有50%几率患病;如果总得分为260,它对应着的患病几率为1,就是一定患病
校准曲线:
cali <- calibrate(lrmModel, method = "boot", B = 1000);
pdf("save_data\\Calibration.pdf", width = 6, height = 6);
plot(
cali,
xlab = "Predicted probability", ylab = "Actual probability",
sub = F
);
dev.off();
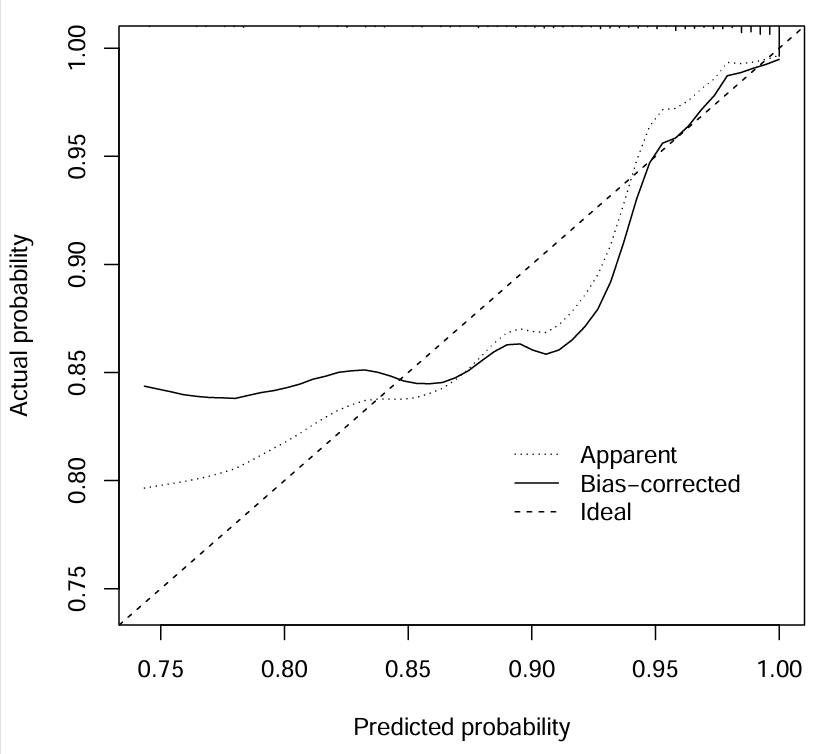
Bias−corrected线与Ideal线(对角线)越重合,结果越好(因为这里是随便选的基因,拟合效果不是很好)
决策曲线,DCA图形:
rt$Type <- ifelse(rt$Type=="Control", 0, 1); # 控制组的值为1
# 决策曲线
dc <- decision_curve(
as.formula(model),
data = rt,
family = binomial(link ='logit'),
thresholds = seq(0,1,by = 0.01),
confidence.intervals = 0.95
);
# DCA图形
pdf(file = "save_data\\DCA.pdf", width = 6, height = 6);
plot_decision_curve(
dc,
curve.names = "six genes",
xlab = "Threshold probability",
cost.benefit.axis = T,
col = "#561215",
confidence.intervals = FALSE,
standardize = FALSE
);
dev.off();
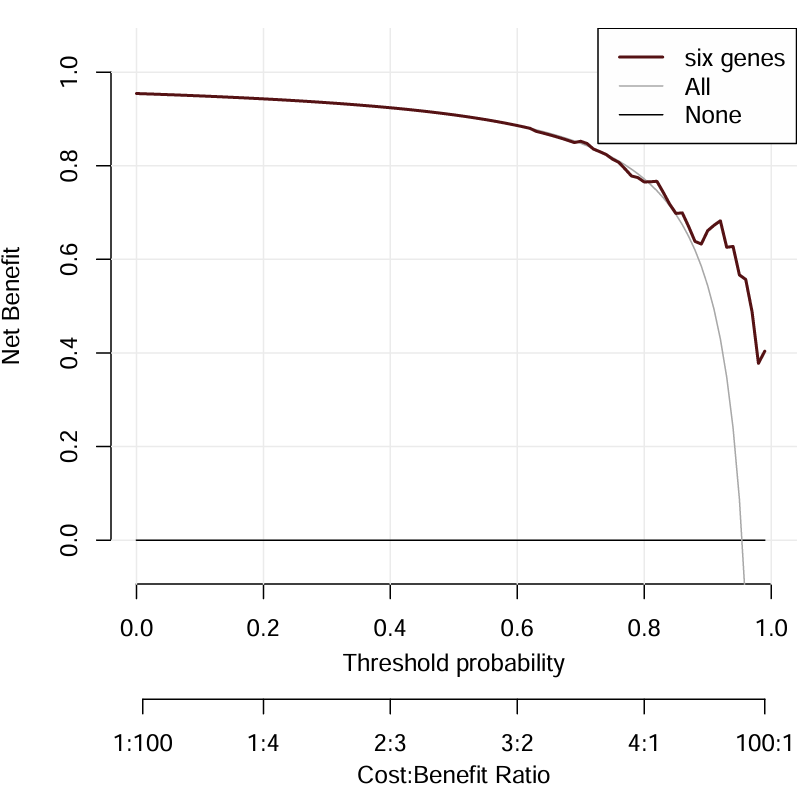
six genes线离All线越远,结果越好
生存状态+风险曲线+表达热图
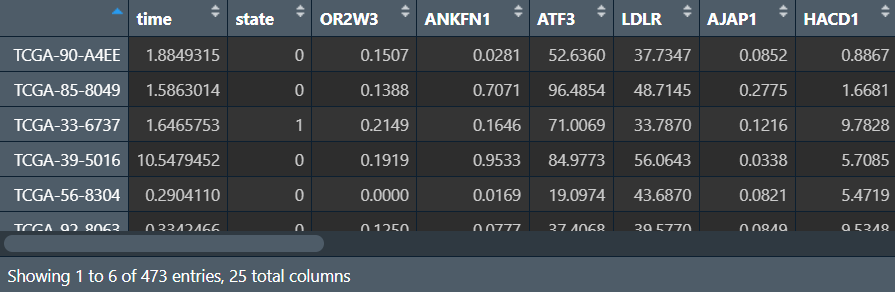
需要数据:风险得分以及各基因在样本中的表达量risk.txt
library(pheatmap);
读取数据,并按风险得分从小到大排序:
rt <- read.table("save_data\\risk.txt", header = T, sep = "\t", check.names = F, row.names = 1);
rt <- rt[order(rt$riskScore), ];
风险曲线:
riskClass <- rt[, "risk"];
lowLength <- length(riskClass[riskClass=="low"]);
highLength <- length(riskClass[riskClass=="high"]);
lowMax <- max(rt$riskScore[riskClass=="low"]); # 高/低风险组分界点的纵坐标
line <- rt[, "riskScore"];
line[line>10] <- 10; # 将超过10的风险得分设置为10
pdf(file = "save_data\\riskScore1.pdf", width = 7, height = 4);
plot(
line,
type = "p",
pch = 20,
xlab = "Patients (increasing risk socre)", ylab = "Risk score",
col = c(
rep("MediumSeaGreen", lowLength),
rep("Firebrick3", highLength)
)
);
abline(h = lowMax,v = lowLength, lty = 2); # 高/低风险组分界线
legend(
"topleft",
c("High risk", "Low risk"),
bty = "n",
pch = 19,
col = c("Firebrick3", "MediumSeaGreen"),
cex = 1.2
);
dev.off();
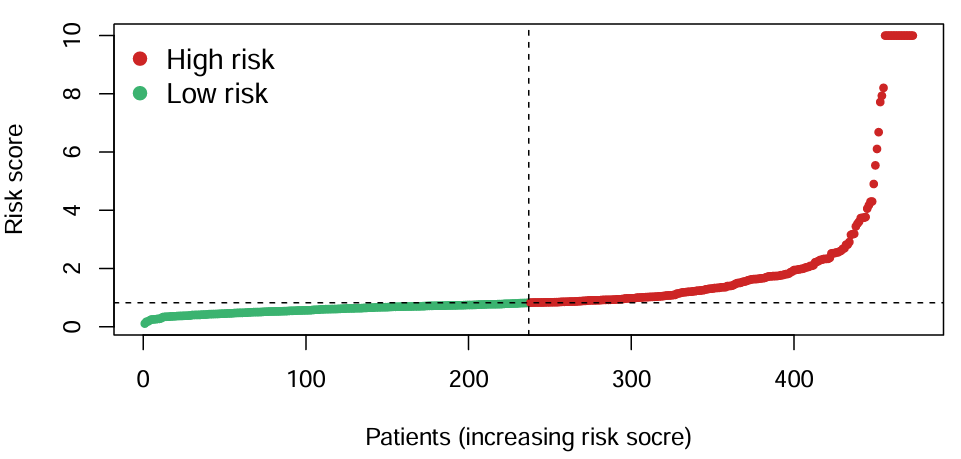
横坐标是病人编号,纵坐标是风险得分,不同颜色代表高/低风险组。可以看到以x轴中点为分界线,一半病人是高风险,一半病人是低风险,右上角点密集处就是风险得分超过10的病人(为了让图不过高,让超过10的风险得分全画在了10的位置上)
生存状态图:
color <- as.vector(rt$state);
color[color==1] <- "Firebrick3";
color[color==0] <- "MediumSeaGreen";
pdf(file = "save_data\\survStat1.pdf", width = 7, height = 4);
plot(
rt$time,
pch = 19,
xlab = "Patients (increasing risk socre)", ylab = "Survival time (years)",
col = color
);
legend(
"topleft",
c("Dead", "Alive"),
bty = "n",
pch = 19,
col = c("Firebrick3", "MediumSeaGreen"),
cex = 1.2
);
abline(v = lowLength, lty = 2);
dev.off();
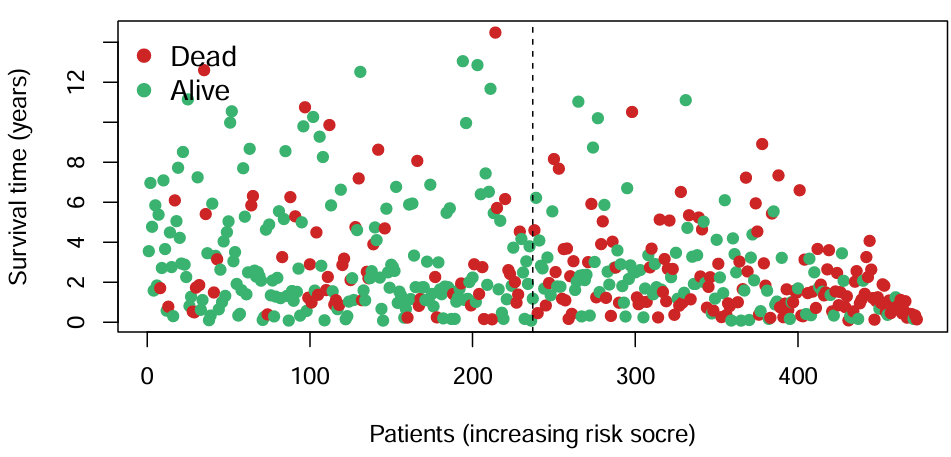
横坐标是病人编号,纵坐标是生存时间/年,不同颜色代表生存状态
热图:
# 颜色
ann_colors <- list();
bioCol <- c("MediumSeaGreen", "Firebrick3");
names(bioCol) <- c("low", "high");
ann_colors[["Risk"]] <- bioCol;
# 画图
rt1 <- rt[c(3:(ncol(rt)-2))]; # 取表达量
rt1 <- t(rt1);
annotation <- data.frame(Risk = rt[, ncol(rt)]);
rownames(annotation) <- rownames(rt);
pdf(file = "save_data\\surv_heatmap1.pdf", width = 6, height = 5);
pheatmap(
rt1,
annotation = annotation,
annotation_colors = ann_colors,
cluster_cols = FALSE,
cluster_rows = FALSE,
show_colnames = F,
scale = "row",
color = colorRampPalette(
c(
rep("MediumSeaGreen", 3.5),
"white",
rep("Firebrick3", 3.5)
)
)(50),
fontsize_col = 3,
fontsize = 7,
fontsize_row = 8
);
dev.off();
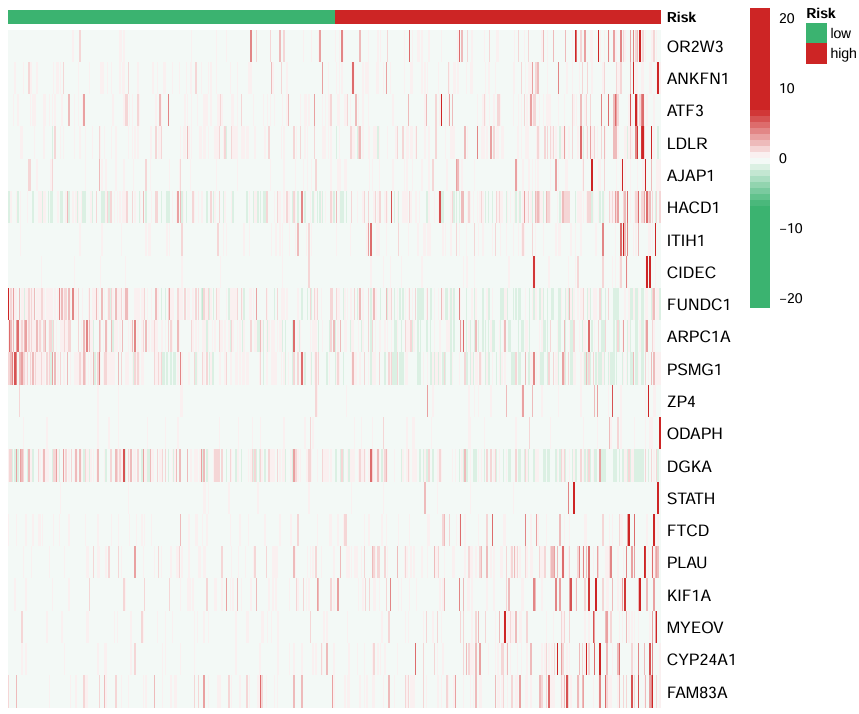
上面的risk标明了高/低风险组,横坐标是样本,纵坐标是基因名,每个块的值是表达量
预测microRNA下游靶基因
if(!require("multiMiR", quietly = T))
{
library("BiocManager");
BiocManager::install("multiMiR");
}
library("multiMiR");
library(tidyverse);
读取mirna文件(获取要预测的mirna名称),进行预测:
# 读取文件
data <- read.table("save_data\\miRNA.RPM.txt", sep = "\t", check.names = F,header = T, row.names = 1);
gene <- rownames(data)[1:5]; # 以前5个mirna为例
example <- get_multimir(
org = "hsa", # 物种:hsa人类、mmu小鼠、rno兔子
mirna = gene, # 要预测的mirna
table = "all",
summary = TRUE
);
example_result <- example@data;
# 保存
write.table(example_result, file = "save_data\\mirna_all.predict.txt", sep = "\t", quote = F, row.names = F, col.names = T);
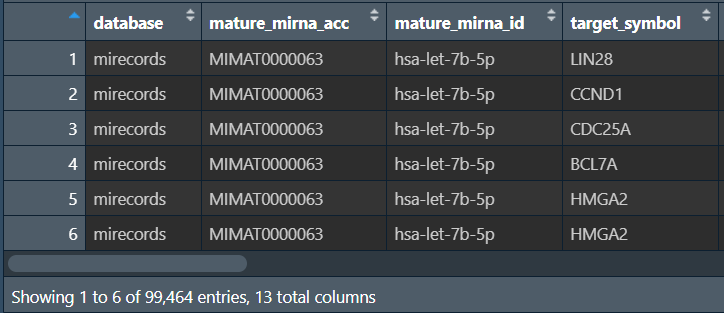
-
mature_mirna_idmirna名称 -
database来自哪个数据库 -
target_symbol下游靶基因 -
type预测方式-
validated实验验证 -
predicted计算机预测 -
disease.drug疾病-药物
-
筛选并去重:
# 展示diana_microt数据库中hsa-let-7a-5p的预测结果
target1 <- example_result %>%
filter(
database == "diana_microt",
mature_mirna_id == "hsa-let-7a-5p"
);
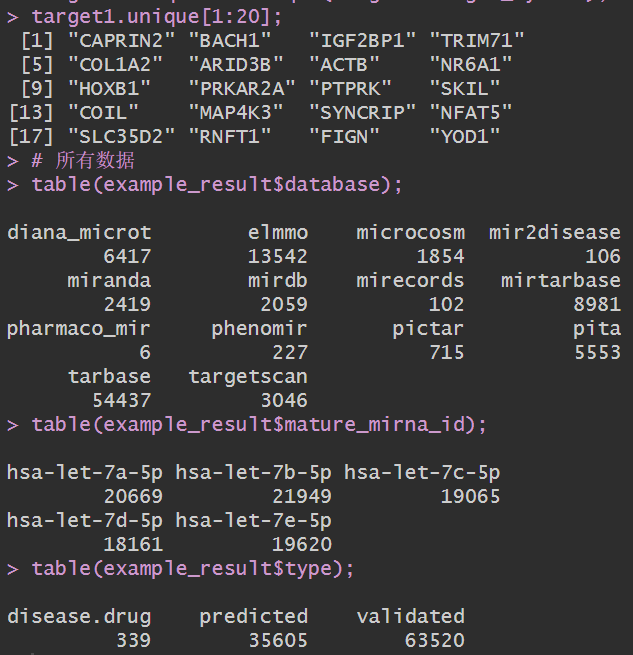
target1.unique <- unique(target1$target_symbol);
target1.unique[1:20];
# 所有数据
table(example_result$database);
table(example_result$mature_mirna_id);
table(example_result$type);
GEO数据库–表达矩阵的间接下载和整理
进入GEO官网,搜索GSE98422,下载Series Matrix File(s)中的两个文件。之前的这个文件都包含表达矩阵,但这个数据集没有(因为文件大小只有几K)
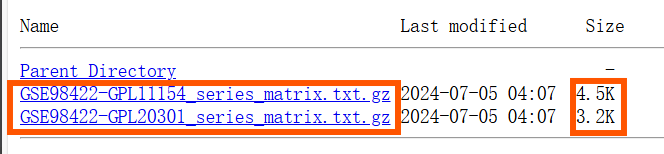
如何找到表达矩阵:(两种方法)
-
将两个
.txt.gz文件解压,打开,找到!Sample_supplementary_file_1的行,根据其中的网址下载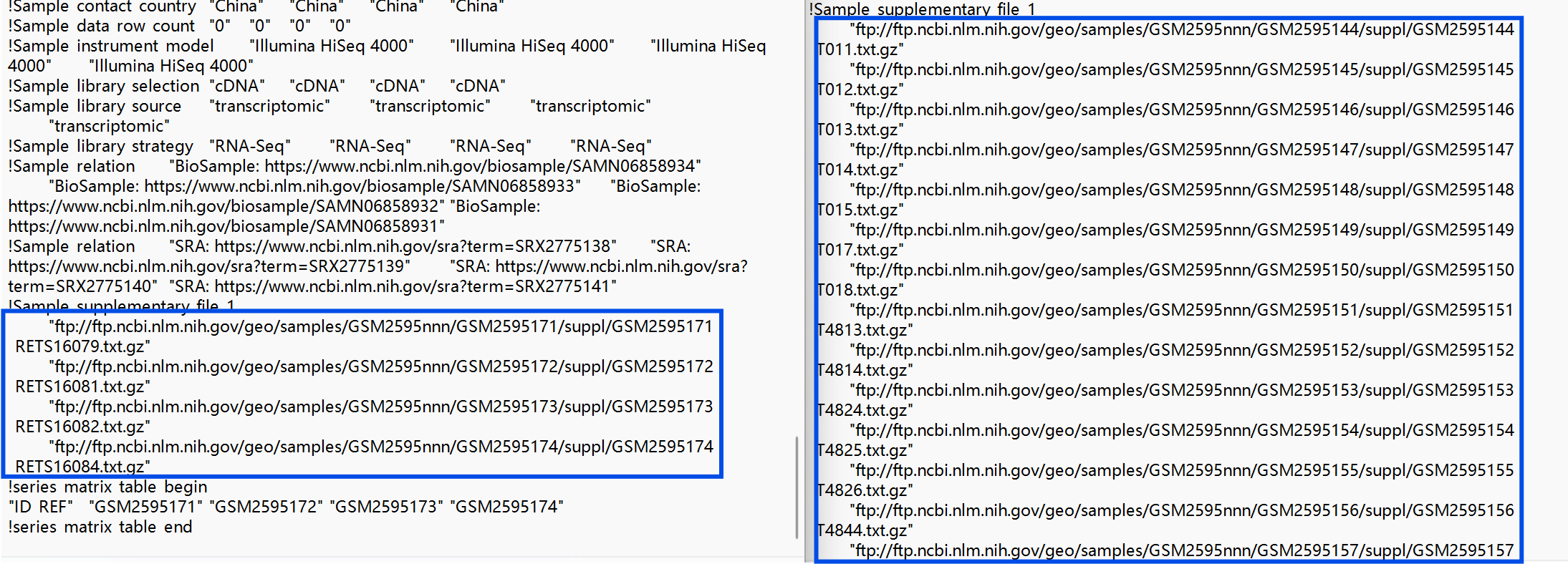
-
在GEO官网中GSE98422的页面,下载GSE98422_RAW.tar(推荐)
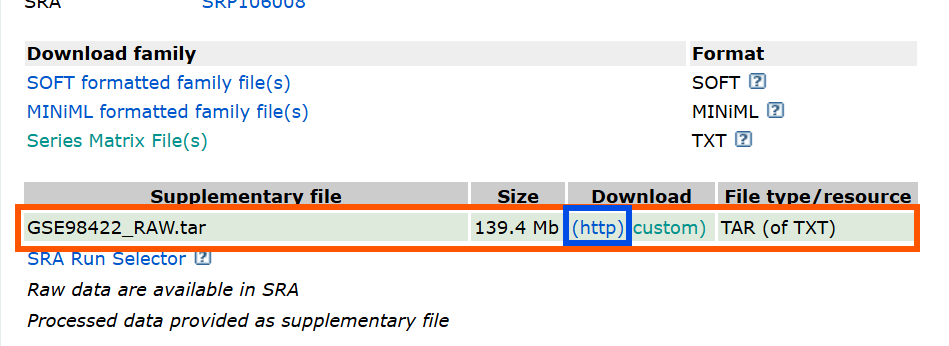
-
http是全部下载 -
custom是选取部分下载(点一下它,下面会出现一个列表,可以进行选取)
下载好后解压成文件夹,其中的数据与第一种方法下载的相同
样本的临床信息:储存在上面提到的两个.txt.gz中,最好将这3个文件都下载
library(GEOquery);
library(stringr);
临床信息:
gset <- getGEO(
"GSE98422",
destdir = "data\\GSE98422",
AnnotGPL = F,
getGPL = F
);
#获取临床信息
pd <- pData(gset[[1]]);
pd2 <- pData(gset[[2]]);
# 保存
write.csv(pd, 'data\\GSE98422\\clinicalGSE98422.1.csv',row.names = TRUE);
write.csv(pd2, 'data\\GSE98422\\clinicalGSE98422.2.csv', row.names = TRUE);
注:这两个临床信息有部分列不同,无法直接合并
表达矩阵:
# 获取全部的表达矩阵文件名
count_file <- list.files('data\\GSE98422\\GSE98422_RAW\\', pattern = '*.txt', recursive = T);
# 看看有多少行
test_data <- read.delim(
paste0('data\\GSE98422\\GSE98422_RAW\\', count_file[1]),
fill = TRUE, header = T, row.names = 1
);
# 结果矩阵
matrix <- data.frame(matrix(nrow = nrow(test_data), ncol = 0));
# 逐个读取及合并
for (i in 1:length(count_file)){
path <- paste0('data\\GSE98422\\GSE98422_RAW\\', count_file[i]);
data <- read.delim(path, fill = T, header = T, row.names = 1);
data <- data[, 4, drop=F];
matrix <- cbind(matrix, data);
}
每个表达矩阵:
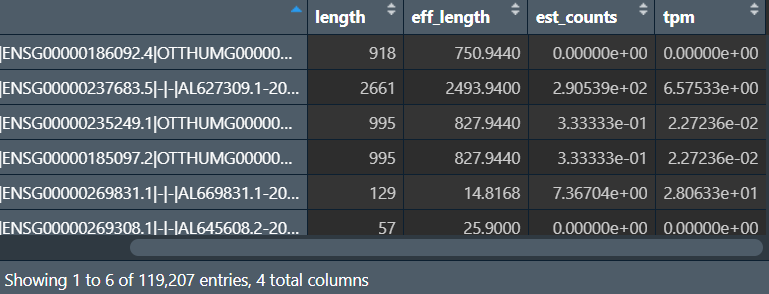
行名是基因id/symbol,est_counts是count值,这里我们获取最后一列tpm值作为表达量
总表达矩阵:
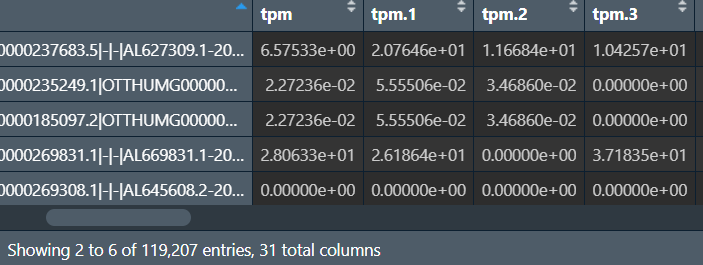
获取样本名,将其作为列名:就是count_file中.txt.gz前面的部分
sample_ids <- c();
for (i in 1:length(count_file)) {
sample_id <- strsplit(count_file, "_")[[i]][1];
sample_ids <- c(sample_ids, sample_id);
}
colnames(matrix) <- sample_ids;
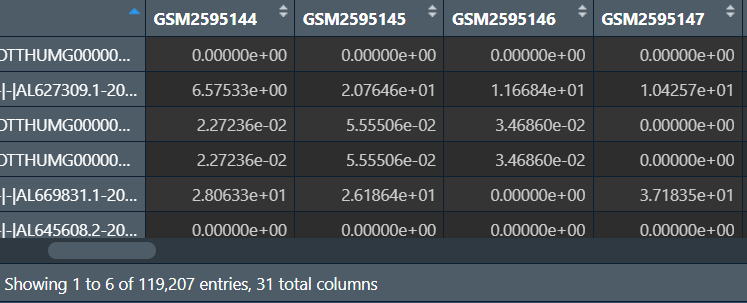
更改基因名(列名):
"ENST00000335137.3|ENSG00000186092.4|OTTHUMG00000001094.1|OTTHUMT00000003223.1|OR4F5-001|OR4F5|918|CDS:1-918|"中OR4F5是基因名(以|分隔的第6项)
matrix$ids <- trimws(
str_split(
rownames(matrix),
'\\|',
simplify = T
)[, 6]
);
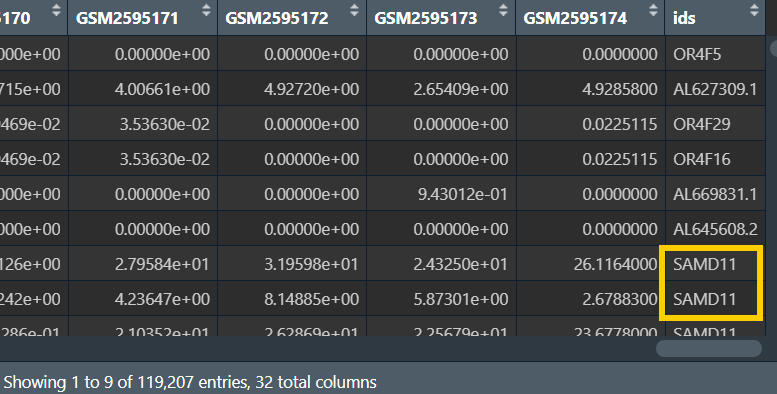
为什么新添加一列ids:因为有些基因名重复,重复值无法直接作行名,需要进行去重
去重(保留最大值):
matrix1 <- aggregate(. ~ ids, data = matrix, max);
# 更改行名为基因名,并删除ids列
rownames(matrix1) <- matrix1[, 1];
matrix1 <- matrix1[, -1];
# 保存
write.table(
data.frame(ID = rownames(matrix1), matrix1),
file = "data\\GSE98422\\GSE98422.txt",
sep = "\t", quote = F, row.names = F
);
TCGA数据库–大于5G的数据下载
进入TCGA官网
以乳腺癌的转录组表达数据为例:
Cohort Builder:
-
Program–TCGA -
Project–TCGA-BRCA
Repository:
-
Data Category–transcriptome profiling -
Data Type–Gene Expression Quantification
Add All Files to Cart加数据添加到仓库,共1231个,5.22GB
下载三个文件:manifest、metadata和cart
下载cart时会弹出提示:
第一种方法(不推荐,下载速度慢,很费时间):使用它推荐的Data Transfer Tool(gdc-client.exe)下载
下载好后将它添加到环境变量中,终端输入命令gdc-client -h查看是否添加完成
之后将下载的manifest文件复制到和它的同目录下,在该目录中打开终端,运行命令:
gdc-client.exe download -m gdc_manifest.2024-03-01.txt
其中gdc_manifest.2024-03-01.txt是manifest文件名,可能需要修改
第二种方法:使用r获取样本名,之后分批次下载
只需下载metadata文件即可,之后运行r代码:
library(rjson);
library(tidyverse);
# 读入metadata文件
json <- jsonlite::fromJSON("data\\大于5G的数据下载\\metadata.cart.2024-03-01.json");
# 获取样本名称及文件名称
sample_id <- sapply(json$associated_entities, function(x){x[, 1]});
file_sample <- data.frame(sample_id, file_name = json$file_name);
# 将样本拆成两组,分别包含615和616个样本
file_sample1 <- file_sample[1:(nrow(file_sample)/2), ];
file_sample2 <- file_sample[(nrow(file_sample)/2):nrow(file_sample)+1, ];
# 导出
write.table(file_sample1, 'data\\大于5G的数据下载\\samples1.txt', sep = "\t", quote = F, row.names = F);
write.table(file_sample2, 'data\\大于5G的数据下载\\samples2.txt', sep = "\t", quote = F, row.names = F);
file_sample2:
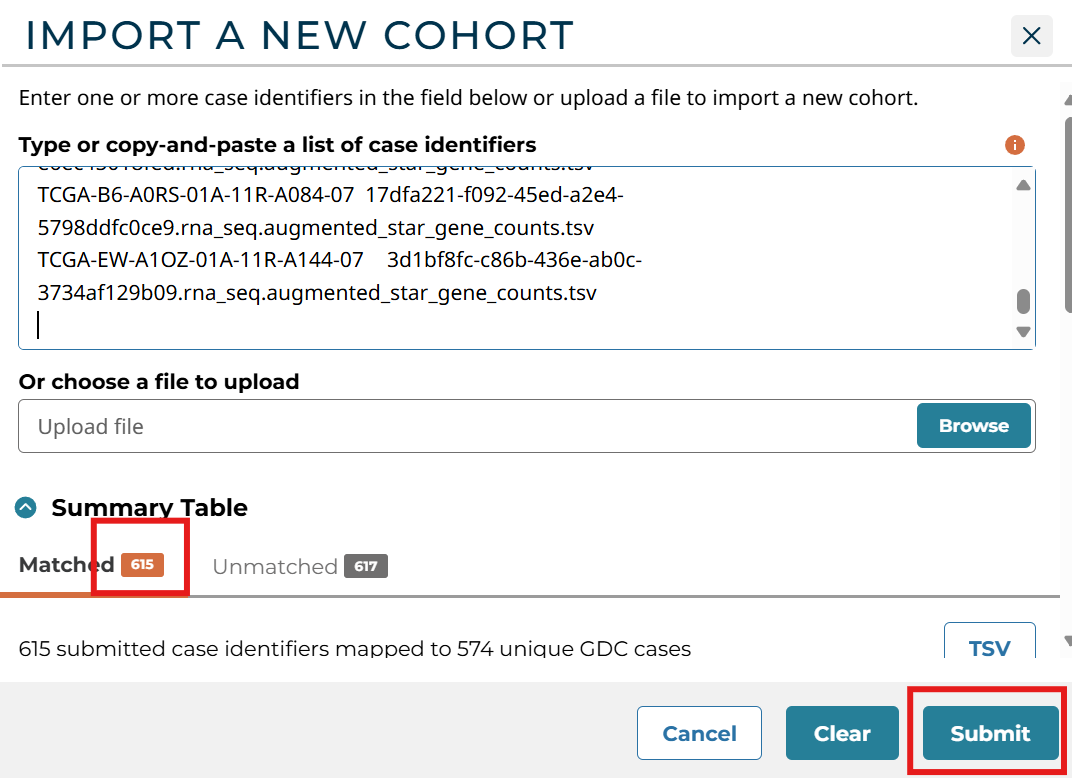
回到TCGA网页中,点击Import New Cohort
将samples1.txt中内容复制粘贴上去,可以看到识别到了615个样本,点击Submit
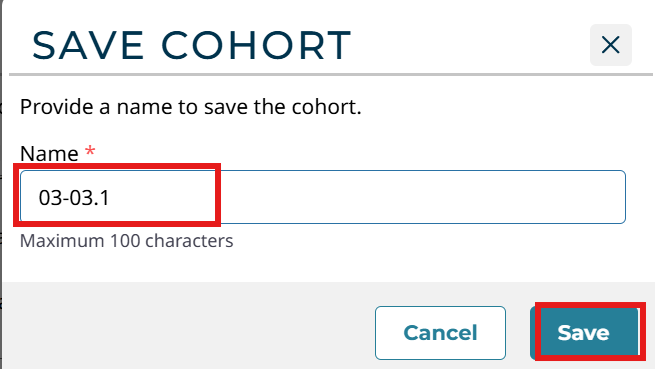
取一个名字,这里我取得是03-03.1,点击Save
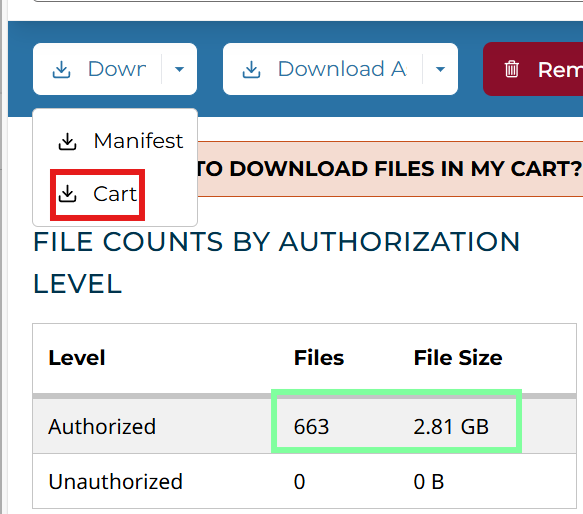
在Repository中筛选:
-
Data Category–transcriptome profiling -
Data Type–Gene Expression Quantification
添加到Cart中,这里只需下载cart文件即可
再同样操作下载samples2.txt,取名为03-03.2
将下载的两个.tar.gz文件解压,将其中的样本文件夹都集中到一个文件夹内,就与之前一般的数据下载得到的结果相同了(过程中提示有相同的文件名,替换/跳过都可)
共有1232个文件(多了一个MANIFEST.txt),正好与之前的1231个文件对应
之后的处理方式同前:
# 读取文件名
count_file <- list.files(
'data\\大于5G的数据下载\\file1\\',
pattern = '*.tsv',recursive = TRUE
);
# 获取每个文件名称
count_file_name <- strsplit(count_file, split = '/');
count_file_name <- sapply(count_file_name, function(x){x[2]});
# 结果矩阵
test_data <- read.delim(paste0('data\\大于5G的数据下载\\file1\\', count_file[1]), fill = TRUE, header = FALSE, row.names = 1);
matrix <- data.frame(matrix(nrow = nrow(test_data), ncol = 0));
# 逐个读取及合并
for (i in 1:length(count_file)){
path <- paste0('data\\大于5G的数据下载\\file1\\', count_file[i]);
data <- read.delim(path, fill = TRUE,header = FALSE, row.names = 1);
colnames(data) <- data[2, ];
data <- data[6]; # 3-unstranded_counts 4-stranded_first 5-stranded_second 6-tpm_unstranded 7-fpkm_unstranded 8-fpkm_uq_unstranded
colnames(data) <- file_sample$sample_id[which(file_sample$file_name==count_file_name[i])];
matrix <- cbind(matrix, data);
}
matrix <- matrix[-c(1:6), ]; # 去除前6行(空行)
# 转化为gene_symbol
test_data <- as.matrix(test_data);
gene_name <- test_data[-c(1:6), 1];
matrix0 <- cbind(gene_name, matrix);
# 获取基因类型
gene_type <- test_data[-c(1:6), 2];
matrix0 <- cbind(gene_type, matrix0);
# 将gene_name列去除重复的基因,保留基因最大表达量结果
matrix0 <- aggregate(. ~ gene_name, data = matrix0, max);
# 只保留mRNA
matrix0 <- subset(x = matrix0, gene_type == "protein_coding");
# 将gene_name列设为行名
rownames(matrix0) <- matrix0[, 1];
matrix0 <- matrix0[, -c(1, 2)];
#导出
matrix1 <- data.frame(ID = rownames(matrix0), matrix0);
colnames(matrix1) <- gsub('[.]', '-', colnames(matrix1));
write.table(matrix1,'data\\大于5G的数据下载\\TPM.txt', sep = "\t", quote = F, row.names = F);
单/多因素cox分析筛选预后相关因素
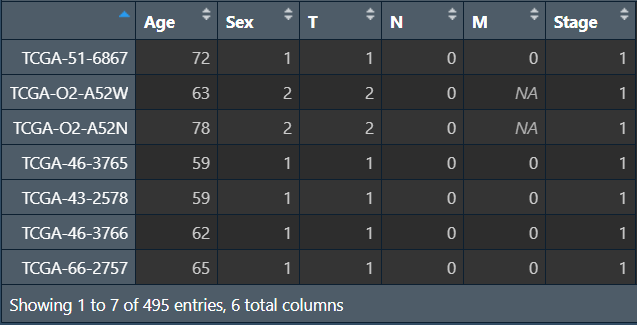
基本方法
需要数据:生存时间/状态、风险得分、患者信息(年龄、性别、TMN分期、stage分期),其中的所有列需要均为数值型(除了样本名),允许空值存在(TMN分期中X表示空值)
注:可以在excel中编辑,编辑好后全选,复制粘贴到一个空txt中,不要把.xlsx直接改成.txt
library(survival);
读取数据:
cli <- read.table("data\\筛选预后相关因素clinical.txt", header = T, sep = "\t", check.names = F, row.names = 1);
risk <- read.table("save_data\\risk.txt", header = T, sep = "\t", check.names = F, row.names = 1);
进行分析:
# 合并两个数据
merge_data <- function(risk, cli){
sameSample <- intersect(row.names(cli), row.names(risk));
risk <- risk[sameSample, ];
cli <- cli[sameSample, ];
rt <- cbind(
time = risk[, 1],
state = risk[, 2],
cli,
riskScore = risk[,(ncol(risk)-1)]
);
}
# 绘制森林图函数
bioForest <- function(coxFile, forestFile = "", forestCol = "#D21E1F"){
rt <- read.table(coxFile, header = T, sep = "\t", check.names = F, row.names = 1);
# 提取数据
gene <- rownames(rt);
hr <- sprintf("%.3f", rt$"HR");
hrLow <- sprintf("%.3f", rt$"HR.95L");
hrHigh <- sprintf("%.3f", rt$"HR.95H");
Hazard.ratio <- paste0(hr, "(", hrLow, "-", hrHigh, ")");
pVal <- ifelse(
rt$pvalue<0.001,
"<0.001",
sprintf("%.3f", rt$pvalue)
);
# 创建图形文件、子图布局
pdf(file = forestFile, width = 6.6, height = 3);
n <- nrow(rt);
nRow <- n+1;
ylim <- c(1, nRow);
layout(matrix(c(1,2), nc = 2), width = c(3, 2.5));
# 左边的临床信息
xlim <- c(0,3);
par(mar = c(4,2.5,2,1));
plot(
1,
xlim = xlim, ylim = ylim,
type = "n", axes = F,
xlab = "", ylab = ""
);
text.cex <- 0.8;
text(0, n:1, gene, adj = 0, cex = text.cex);
text(1.5-0.5*0.2, n:1, pVal, adj = 1, cex = text.cex);
text(1.5-0.5*0.2, n+1, 'pvalue', cex = text.cex, font = 2, adj = 1);
text(3.1, n:1, Hazard.ratio,adj = 1, cex = text.cex);
text(3.1, n+1, 'Hazard ratio', cex = text.cex, font = 2, adj = 1);
# 右边的森林图
par(mar = c(4, 1, 2, 1), mgp = c(2, 0.5, 0));
xlim <- c(0, max(as.numeric(hrLow), as.numeric(hrHigh)));
plot(
1,
xlim = xlim, ylim = ylim,
type = "n", axes = F, xaxs = "i",
ylab = "", xlab = "Hazard ratio"
);
arrows(
as.numeric(hrLow), n:1,
as.numeric(hrHigh), n:1,
angle = 90, code = 3, length = 0.05,
col = "darkblue", lwd = 2.5
);
abline(v = 1, col = "black", lty = 2, lwd = 2);
boxcolor <- ifelse(as.numeric(hr) > 1, forestCol, forestCol);
points(
as.numeric(hr), n:1,
pch = 15, col = boxcolor, cex = 1.5
);
axis(1);
dev.off();
}
# 单因素分析
indep_uniCox <- function(rt, project_path = ""){
uniCoxFile <- paste0(project_path, "indep_uniCox.txt");
uniCoxPdf <- paste0(project_path, "indep_uniCox.pdf");
uniTab <- data.frame();
for(i in colnames(rt[, 3:ncol(rt)])){
cox <- coxph(Surv(time, state) ~ rt[,i], data = rt);
coxSummary <- summary(cox);
uniTab <- rbind(
uniTab,
cbind(
id = i,
HR = coxSummary$conf.int[, "exp(coef)"],
HR.95L = coxSummary$conf.int[, "lower .95"],
HR.95H = coxSummary$conf.int[, "upper .95"],
pvalue = coxSummary$coefficients[, "Pr(>|z|)"]
)
);
}
# 保存数据
write.table(uniTab, file = uniCoxFile, sep = "\t", row.names = F, quote = F);
# 绘图
bioForest(uniCoxFile, forestFile = uniCoxPdf, forestCol = "#1E4A93");
return(uniTab);
}
# 多因素分析
indep_multiCox <- function(rt, uniTab, project_path = ""){
# 多因素独立预后分析
multiCoxFile <- paste0(project_path, "indep_multiCox.txt");
multiCoxPdf <- paste0(project_path, "indep_multiCox.pdf");
# 是否纳入小于0.05的参数--更改<1的筛选条件为<0.05即可
uniTab <- uniTab[as.numeric(uniTab[, "pvalue"])<1, ];
rt1 <- rt[,c("time", "state", as.vector(uniTab[, "id"]))];
multiCox <- coxph(Surv(time, state) ~ ., data = rt1);
multiCoxSum <- summary(multiCox);
multiTab <- data.frame();
multiTab <- cbind(
HR = multiCoxSum$conf.int[, "exp(coef)"],
HR.95L = multiCoxSum$conf.int[, "lower .95"],
HR.95H = multiCoxSum$conf.int[, "upper .95"],
pvalue = multiCoxSum$coefficients[, "Pr(>|z|)"]
);
multiTab <- cbind(id = row.names(multiTab), multiTab);
# 保存数据
write.table(multiTab, file = multiCoxFile, sep = "\t", row.names = F, quote = F);
# 绘图
bioForest(multiCoxFile, forestFile = multiCoxPdf, forestCol = "#D21E1F");
return(multiTab);
}
rt <- merge_data(risk, cli);
uniTab <- indep_uniCox(rt, "save_data\\");
multiTab <- indep_multiCox(rt, uniTab, "save_data\\");
画图使用的数据:
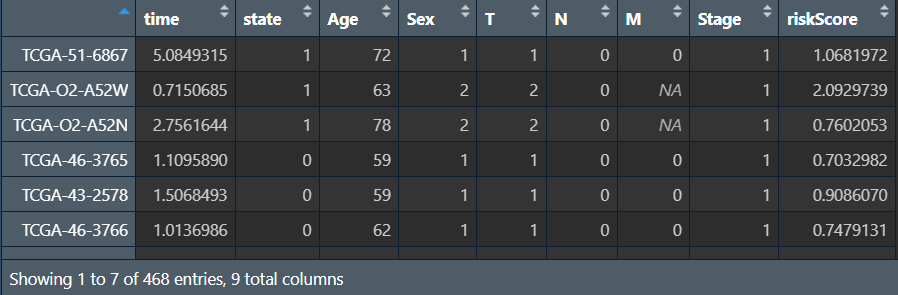
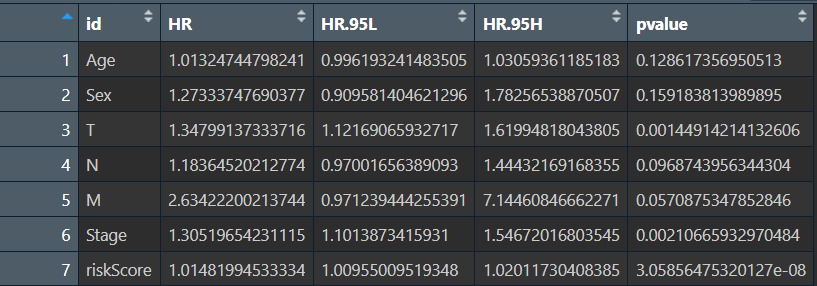
单因素计算结果uniTab:
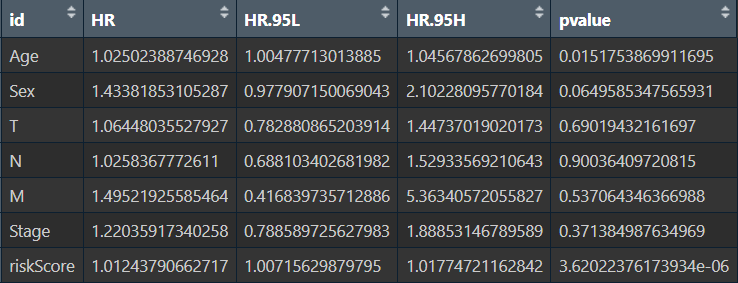
多因素计算结果multiTab:
单因素绘图结果:
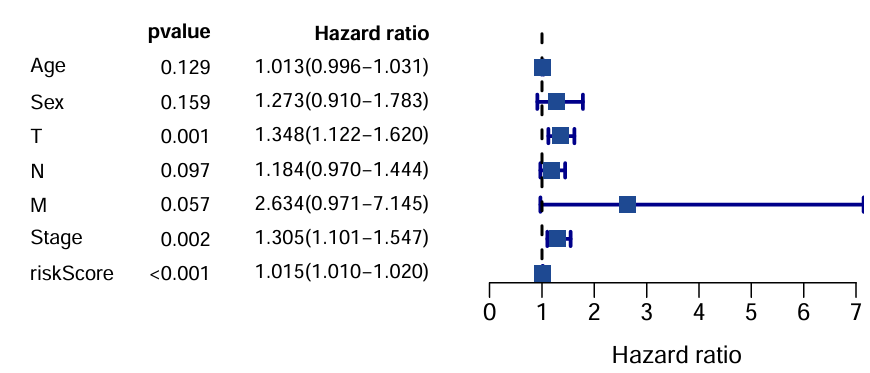
可以看到风险得分、Stage分期、T分期的p值<0.05
多因素绘图结果:
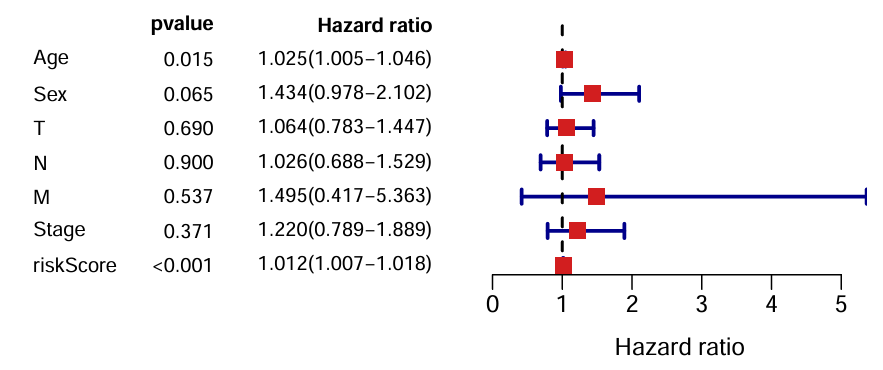
可以看到只有风险得分、年龄的p值<0.05
区别:单因素是看单个因素和预后(生存情况)的关系;多因素是看多个因素,验证的是独立预后因素。单因素结果中某个因素的p<0.05说明它是一个预后因素,如果该因素在多因素分析中p仍<0.05,就说明它是一个独立预后因素
为什么M的HR值跨度大:因为M取值基本都为0,值为1的很少,而且空值也很多,不适合作cox分析
一个问题:性别、TMN、Stage都是离散变量,而Age是连续变量。这种做法实际上把离散变量都当成了连续变量
改进
需要数据:生存时间/状态、风险得分、患者信息(年龄、性别、TN分期、stage分期,没有M列)
删除有空值的行(含X的值也算空值),只有年龄列是数值型,其它均为字符型(T1/N0/FEMALE/Stage II)
if(!require("autoReg", quietly = T))
{
install.packages("autoReg");
}
if(!require("forestplot", quietly = T))
{
install.packages("forestplot");
}
library("autoReg");
library(survminer);
library(survival);
library(forestplot);
读取数据,合并:
# 读取
cli <- read.table("data\\筛选预后相关因素改进clinical.txt", header = T, sep = "\t", check.names = F, row.names = 1);
risk <- read.table("save_data\\risk.txt", header = T, sep = "\t", check.names = F, row.names = 1);
# 合并
sameSample <- intersect(row.names(cli), row.names(risk));
risk <- risk[sameSample, ];
cli <- cli[sameSample, ];
rt <- cbind(
time = risk[, 1],
state = risk[, 2],
cli,
riskScore = risk[, ncol(risk)-1]
);
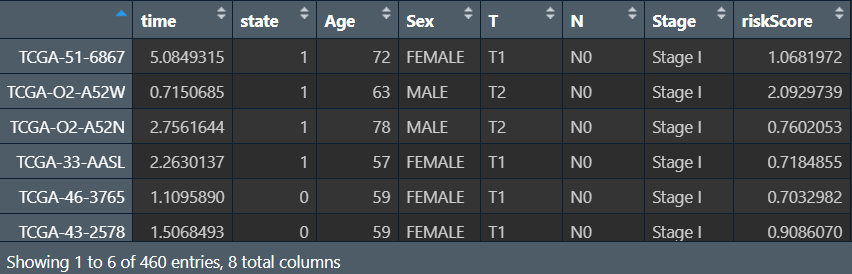
更改列类型,并去除空值:将连续变量(年龄、风险得分)设为数值型,其它离散变量设为factor类型
# 更改列类型
rt[, 3] <- as.numeric(rt[, 3]);
rt[, 4] <- factor(rt[, 4]);
rt[, 5] <- factor(rt[, 5]);
rt[, 6] <- factor(rt[, 6]);
rt[, 7] <- factor(rt[, 7]);
rt[, 8] <- as.numeric(rt[, 8]);
# 删除空值及NA
rt <- rt[apply(rt, 1, function(x){
any(is.na(match('unknow', x)));
}), , drop=F];
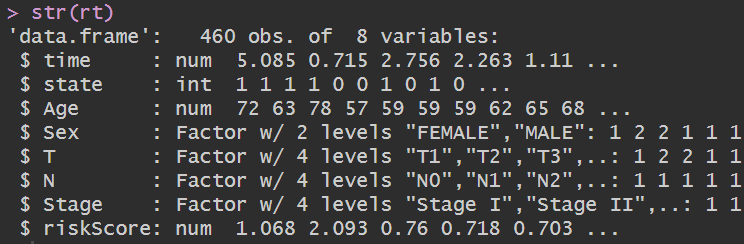
构建cox回归模型:
coxmod <- coxph(Surv(time, state) ~ ., data = rt);
# 筛选,阈值是threshold,设为1就是全部纳入
ft3 <- autoReg(coxmod, uni = TRUE, threshold = 0.05);
# myft(ft3); # 查看结果
# 保存
write.table(ft3, file="save_data\\multicox_result.txt", sep = "\t", row.names = F, quote = F);
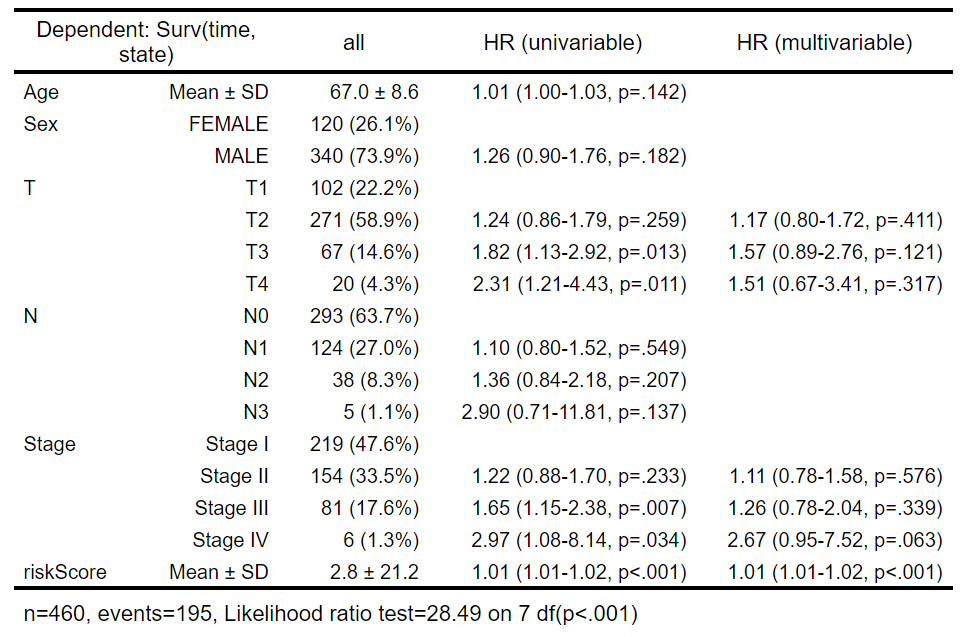
结果中Mean ± SD的就是连续变量,其它列出具体值的是离散变量。没有HR的就是参照值reference,可以看到每个离散变量的第一个值是参照
最终Stage、T、riskScore的p<0.05
多因素cox分析,绘制森林图:
# 选取p<0.05的因素作cox分析
multiCox <- coxph(Surv(time, state) ~ Stage + T + riskScore, data = rt);
multiCoxSum <- summary(multiCox);
multiTab <- data.frame();
multiTab <- cbind(
HR = multiCoxSum$conf.int[, "exp(coef)"],
HR.95L = multiCoxSum$conf.int[, "lower .95"],
HR.95H = multiCoxSum$conf.int[, "upper .95"],
pvalue = multiCoxSum$coefficients[, "Pr(>|z|)"]
);
write.table(cbind(id = row.names(multiTab), multiTab), file = "save_data\\mul.cox.result.txt", sep = "\t", row.names = F, quote = F);
# 绘制森林图
pdf(file = "save_data\\multicox_forest.pdf", width = 8, height = 6, onefile = FALSE);
ggforest(
multiCox,
data = rt,
main = "Hazard ratio",
cpositions = c(0.02, 0.22, 0.4),
fontsize = 0.8,
refLabel = "reference",
noDigits = 3
);
dev.off();
多因素cox分析结果multiTab:
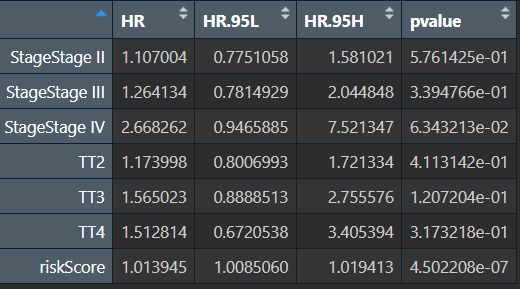
其中HR值与上面ft3的HR (multivariable)是一样的
森林图:
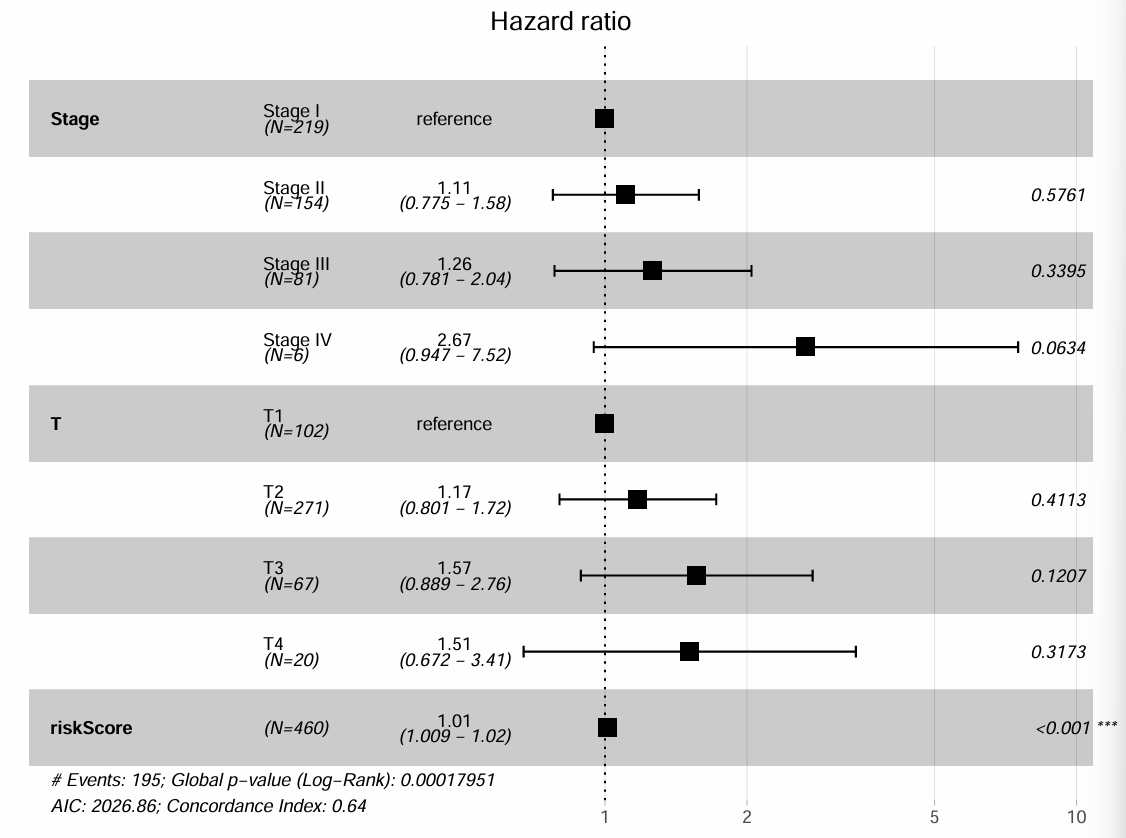
图中的N表示样本数,可以看到样本越少,HR值偏差越大