文献阅读2
other-other2025.9.25-2025.10.17研究进展
ENCODE和UCSC基因组浏览器简介
ENCODE(Encyclopedia of DNA Elements):标准化、高质量的功能组学数据,包括功能元件注释(增强子位置、启动子范围等)、基因表达数据(不同组织各种状态下基因与非编码RNA表达量)、表观遗传数据(组蛋白修饰、DNA甲基化、染色质可及性等)、蛋白质DNA互作数据(转录因子、组蛋白等蛋白在基因组上的结合位点)、三维基因组数据(染色质的空间结构)
- 应用:
- 基因调控机制研究:例如可以查询某基因的上游增强子位置,或特定转录因子的靶基因
- 疾病关联分析:将GWAS发现的SNP定位到ENCODE注释的调控元件上,推测其影响疾病的分子机制,例如SNP破坏增强子与基因的结合
- 数据挖掘与整合:下载某细胞系的ChIP-seq数据(如某蛋白与基因组的结合数据),与自身的RNA-seq数据联合分析,筛选该蛋白调控的差异表达基因
- 数据筛选:物种(Species)+实验类型xxx-seq(Assay Type)+细胞系(Biosample)
- 数据下载:原始测序数据FASTQ、比对结果BAM、峰值文件BED、注释文件GTF
- 同时还有Genome Browser可视化工具,可快速查看某基因组区域的ENCODE注释
- 简单介绍
- CHIP-seq数据下载
UCSC Genome Browser:交互式可视化基因数据,为基因组序列提供直观的可视化注释与多维度数据整合
hERV相关论文
概述
- HERV约占人类基因组的8%,是远古逆转录病毒感染后遗留的整合序列,在演化过程中逐渐被宿主共生利用。完整的ERV含有病毒基因(gag、pol、env)及两端LTR,而多数现代ERV因进化中LTR重组失去内部序列,只剩孤立LTR
- HERV的长末端重复序列可作为启动子或增强子,调控邻近基因表达;HERV的插入还会造成基因组重排或插入突变,影响基因组稳定性;宿主也可通过表观遗传等机制限制其活性
- HERV编码的蛋白参与胎盘发育和胚胎着床等早期发育过程,例如子宫融合蛋白(syncytin)。在人类早期胚胎时期,HERVL/HERVH高度表达,与全基因组激活和细胞命运决定/维持胚胎干细胞多能性有关;在发育过程中,ERV的适度激活为重要基因网络提供调控元件,而失控激活则可能扰乱神经发生等过程
- HERV既是人体免疫监视的一部分,也能在失控时促进疾病:大量HERV插入显著扩展了干扰素调控网络,部分HERV插入位于关键免疫基因附近,影响这些基因的表达和遗传多态性;同时,ERV转录产生的双链RNA或cDNA可被先天免疫受体识别,激活相关信号通路(如I型干扰素通路),这种“类病毒效应”在宿主抗感染中发挥作用,例如活化的ERV反转录酶可裂解入侵病毒的RNA,细胞在不良状态时可诱导ERV表达,进而提高细胞免疫状态;HERV Env蛋白的免疫抑制功能使其在正常生理状态下帮助抵御过度炎症,但当HERV异常激活时也可能诱发自身免疫
- 衰老组织中ERV表达上升可能导致慢性炎症,与神经退行性疾病和多种衰老相关病理过程相关。HERV-K在衰老细胞中高度表达,可利用针对HERV-K的T细胞来清除衰老细胞,从而缓解衰老相关损伤
- HERV-K(HML-2)家族的致病性:HERV-K位点多态性与肿瘤发生和自身免疫性疾病的个体间差异有关,可能是遗传致病因素——HERV-K的Env蛋白可介导细胞膜融合,并激发宿主的先天和适应性免疫反应;HERV-K dUTP酶可诱导炎症性细胞因子产生,与银屑病等自身炎症疾病相关。目前无确凿证据证明HERV-K直接导致疾病,但发现其在多种癌症和自身免疫性疾病病变组织中高度表达,因此其表达产物可成为免疫监视的靶点;同时它在某些正常组织中也有低水平表达,说明其可能具有潜在生理功能。HERV-K的多态性插入位点可能解释个体间的疾病易感差异
- 在肿瘤治疗中的一些临床应用:将HERV蛋白制成疫苗,诱导强烈T细胞反应,进而抑制肿瘤生长;HERV-W Env的单克隆抗体可以减弱HERV引发的致炎反应;通过使用去甲基化药物等方法,解除肿瘤细胞中HERV沉默,模拟病毒感染,提高免疫检查点抑制剂的疗效
详细的数据分析方法
- 阿尔茨海默病和额颞叶痴呆患者组织中内源性逆转录病毒的分析:收集gAD/sAD与gFTD/sFTD患者以及对照者的临床样本(外周血样本和前额叶皮质脑组织),进行qRT-PCR(定量逆转录PCR:结合了逆转录和实时定量PCR的过程,能够在PCR扩增过程中实时监测产物的量)定量不同hERV家族的转录本水平,之后以常见的内参基因(actin、GAPDH等)进行标准化,比较健康组与患者组组间差异,发现HERV-K env在sAD患者血液中显著上调;对部分样本进行Infinium MethylationEPIC微阵列分析,检测全基因组DNA甲基化水平,发现一些参与hERV调控的基因(如所有组中的APOBEC3、E2F5,和FTD中的CDK6、PRODH、SLC4A8)呈现高甲基化
- HERV-K env转录本可作为一种潜在的临床生物标志物,未来可对env蛋白进行更多研究;参与hERV调节机制的基因在AD/FTD脑组织中存在表观遗传差异,说明不同神经退行性疾病中hERV激活可能涉及不同机制,可以作为不同神经疾病的区分标志;同时,对这些差异基因的研究可能揭示导致HERV激活的潜在生物学机制
- 与胶质母细胞瘤风险和预后相关的人类内源性逆转录病毒:使用TCGA中的RNA-Seq数据(包含胶质母细胞瘤GBM、低级别胶质瘤LGG和正常脑组织NB的样本),使用Telescope工具进行hERV定位特异性的表达定量(先使用SAMtools将BAM格式转成FASTQ格式,再使用Bowtie2将RNA-Seq数据比对到参考基因组上,最后使用Telescope相关文献中给出的hERV注释计算每个hERV位点的表达量),使用DESeq2基于负二项分布和方差稳定变换进行差异表达分析。筛选出表达差异较大的hERV后,对它们进行PCA分析以再次减少关键基因数量,之后构建了胶质瘤特异性HERV得分(GH Score),进行KM生存分析,并采用对数秩检验和Cox回归模型评估GH得分与生存的相关性
- 功能预测:hERV基因组区段缺乏直接功能注释,因此使用GREAT工具将每个hERV片段与其潜在调控的基因关联,使用GeneMANIA构建由这些基因组成的功能关联网络,并通过QIAGEN IPA对上述关联基因进行通路富集分析
- 在染色体图谱上标记与GBM密切相关的hERV位置,构建了胶质瘤全基因组范围的hERV转录组图谱,确定了与肿瘤级别和生存预后相关的hERV标志(GH Score);通过功能预测发现这些差异表达hERV与18个电压门控钾通道基因密切相关,提示hERV可能通过调控电生理通路参与GBM的发生发展
- 基于ENCODE RNA-seq数据的人内源性逆转录病毒差异表达分析:利用ENCODE项目的Tier 1/2细胞系RNA-seq数据,对12种细胞系(6个正常6个癌症)中的各HERV位点和HERV家族表达进行综合差异分析。HERV基因组位置信息来源于HERVd数据库,使用liftOver将HERVd基于旧版hg17基因组的坐标转换到hg19,通过BLAT比对找回了在新版本中缺失或被拆开的序列,并在hg19中搜索了HERVd未标注的区域以发现新增的内源病毒元件;使用FeatureCounts统计了每个样本中比对到上述HERV坐标的读段数量,得到各样本的HERV计数矩阵;对计数进行库大小标准化,并采用方差稳定变换处理(减小技术噪声和不同样本间变异),用DESeq对不同细胞条件进行差异表达分析(对单端/双端测序数据进行PCA分析,发现差异显著,因此在差异分析中剔除了单端数据,只保留各细胞系的双端测序结果);对结果做了PCA分析,检测所有样本HERV表达模式的全局差异(区分正常-癌细胞);将每种细胞类型中上调的HERV位点按家族归类,以统计活跃HERV家族在不同细胞的富集情况。最后还对管家基因重复上述分析,发现覆盖率评估都与HERVd完全相同,说明HERV分析结果不是测序或计算偏差所致
- 补充——Tier 1/2细胞系:Tier 1是ENCODE项目中优先级最高、数据最全面的细胞系,数量较少,涵盖几乎所有实验类型(RNA-seq、ChIP-seq、Exon Arrays等)并设置至少2个生物学重复;Tier 2包含更多样化的细胞类型/组织来源/细胞状态,能满足更细分的研究需求,实验覆盖范围略少于1。Tier 1作为通用参考,Tier 2作为更多场景的补充,使ENCODE数据既能支撑基础通用研究,又能覆盖不同组织、疾病状态的细胞特性
- 补充——FeatureCounts计数时仅统计每个read的主要比对位置,从而避免将无法唯一定位的reads重复计入:有些read过于普通,可能匹配多个HERV位点,尤其是新的HERV家族(进入人类基因组没多久,复制出来的版本还很相似,更容易造成混淆),如果这些read被重复算到多个HERV上,就会把这些HERV位点的表达量提高。因此规定每个read只算一次,只算到匹配度最高的那个比对位置(primary alignment),可能会漏掉一些信息,但能避免重复加分,减少误差
结果:
- PCA显示,6个正常细胞与6个癌细胞在HERV表达空间中基本可以区分,说明总体上肿瘤细胞的HERV表达模式有别于正常细胞,但对显著差异的HERV进行家族归类后,没有发现单一HERV家族能统一地区分正常组和癌症组,说明肿瘤涉及的表达变化来自多个HERV家族。但仍发现了某些HERV家族在部分细胞系中出现高表达,例如ERVL和HERV-L家族,以及人类中最年轻、具有高反转录活性的HERVK,和在胚胎干细胞中高度表达、被认为是多能性标志的HERVH,说明不同细胞类型在HERV活跃家族上有一定特征模式==>HERV表达谱的综合分析能揭示细胞的疾病状态及潜在异常
- 存在两个异常行为的细胞样本——CSHL的GM12878在实验设计中属于正常组,但被聚类到肿瘤组,说明该细胞系可能发生了部分转化或实验处理差异导致偏差;Caltech的HeLa-S3表现出异常的HERVH高表达,说明该细胞系可能具有未分化或返祖的分子特征==>HERV表达可用于发现潜在的细胞状态偏差,HERVK/HERVH可能具有促瘤作用,有可能作为癌症的生物标记物或治疗靶点
- 除上述特例外,大部分相同细胞系的不同批次样本HERV表达高度相似,对管家基因的重复分析证明ENCODE提供的不同批次数据经过适当处理后可用于一致的HERV分析
局限性:舍弃了单端测序样本以保证分析的一致性,有效样本量有所降低;研究对象仅限于细胞系,而不是患者原始肿瘤组织或正常组织,细胞系长期体外培养可能产生特殊的HERV表达模式;主要基于统计分析,没有研究对于某些HERV在特定细胞中高表达的生物学机制
- EnHERV:特定人类内源性逆转录病毒模式及其邻近基因的富集分析:采用基于Repbase的RepeatMasker对来自UCSC的人类hg19参考基因组进行全基因组扫描,初步识别HERV相关的LTR序列和内部元件;由于大量HERV由于插入/缺失而被分段注释,使用REannotate按照片段间距离、方向和家族归属对RepeatMasker输出进行聚合,将属于同一HERV元件的碎片合并为单一注释记录;将结果整合为可供其他用户使用的EnHERV数据库,具有搜索功能(按基因/HERV特征搜索)和富集分析功能——比较给定基因列表中“含特定HERV特征的基因”与“不含该特征的基因”的比例、和全基因组背景下相应比例的差异,之后采用Fisher精确检验判断某条件下的差异基因集与特定HERV特征是否存在显著富集关联(具体来说,上调基因中含该特征/不含该特征、全基因组背景中含该特征/不含该特征,如果这两个比值有显著差异,就说明这种HERV特征在该疾病的上调或下调基因中“过度出现”,可能具有生物学作用)
结果:
- 通过EnHERV的关联富集分析,“基因内HERV”与基因表达上/下调在多种疾病中相关,而在不同疾病类型中呈现相反趋势,例如在大多数癌症以及银屑病,携带基因内HERV的基因显著倾向于下调表达(与其他转座元件的作用类似,癌细胞DNA去甲基化导致转座元件异常激活,其反义转录干扰基因转录),说明HERV谱系的异常可作为癌症表观遗传失调的标志;而在自身免疫疾病的免疫细胞中,基因内HERV的存在与基因上调显著相关(异常激活的HERV及其转录本可能通过模拟病毒成分刺激免疫受体,从而过度激活免疫通路),说明可通过阻断这些HERV转录或产物以减轻自身免疫反应
- 对于LTR序列,一些HERV家族在特定条件(疾病)下表现突出
- HERV插入在基因组中的方向偏性,HERV与宿主基因呈反向排列的约占54.05%,略高于同向排列
- ==>HERV在不同病理状态下表现出特异的关联模式,在癌症和自身免疫等疾病中具有潜在功能作用
局限性:
- 富集分析揭示的只是相关性,并不能证明HERV插入直接导致了基因表达改变。例如基因内HERV与下调基因相关,可能是HERV插入干扰转录所致,也可能反映这些基因附近富集的HERV同时受到整体异染色质化影响
- 未直接测量HERV本身的转录水平,不知道HERV是否被转录以及转录产物对基因表达的影响
- 将人类内源性逆转录病毒整合到转录组范围内的关联研究中,突显了主要精神疾病的新风险因素:在TWAS中引入HERV表达,以评估HERV转录水平是否与精神疾病的遗传风险相关。证实了成年人大脑皮质中存在大量HERV转录本的表达,相当一部分HERV表达具有可遗传性,它们在不同个体间的表达差异可由邻近基因组变异来解释,同时不同人群间HERV表达的遗传调控存在差异;通过rTWAS分析,在欧洲裔样本中发现了若干显著的HERV表达-疾病关联信号(针对五种主要精神障碍,共识别出26个HERV表达与疾病易感性存在统计学显著关联);因为许多HERV邻近基因且可能共享遗传信号,因此筛选了独立于周边基因表达且与疾病相关的HERV位点;通过WGCNA分析,观察到HERV可能参与了多种生物学过程(突触功能、线粒体功能、免疫反应、信号转导和刺激感应)
RNA-seq数据(.bam格式)->使用samtools转成FASTQ->Trimmatic质控->Bowtie2将RNA-seq读段比对到人类参考基因组(hg38)->使用Telescope对比对结果进行处理(使用HERV注释量化HERV表达),Telescope通过贝叶斯模型和EM算法将模糊比对的读段重新分配到最可能的HERV来源位点,不简单丢弃有多重匹配的读段,而是判定其最可能的位点,使每个HERV插入位点的表达量得以准确估计- 使用kallisto评估常规蛋白编码基因的表达,从而量化基因表达,以直接比较同一基因座上HERV的表达信号和邻近基因的表达信号,从而判断HERV信号是否独立于邻近基因,同时为后续构建共表达网络、推断HERV可能参与的生物学功能模块提供数据
- 过滤掉低表达的转录本,对表达矩阵进行了标准化和校正——TMM方法归一化、主成分分析检测并移除明显离群样本、线性模型和潜在变量校正调整可能影响表达的协变量(批次、样本性别/年龄/种族等),剔除非遗传因素造成的表达差异
- 为了与公开的GWAS结果相匹配,将HERV和基因的基因组坐标从hg38转换为hg19版本
- 使用FUSION工具构建顺式调控的表达预测模型并执行TWAS分析:对每个转录本,训练一个“用附近SNP预测该转录本表达”的模型,筛选具有显著顺式遗传力的HERV和基因;进行TWAS,检测每个转录本的遗传预测表达水平是否与疾病表型显著相关,在全基因组范围筛选出与疾病易感性相关的表达信号,这些信号可能代表潜在的致病基因或HERV
- Bonferroni校正,控制假阳性;由于TWAS可能检测到同一基因组区域内多个相关联的表达特征(例如某基因与HERV都显著,但可能都是因为同一个遗传变异),引入FUSION的条件分析模块,重新评估其他转录本的关联,以识别条件独立的表达关联;同时使用FOCUS工具对显著的TWAS信号进行精细定位分析,控制相邻表达信号的相关性,识别出在同一遗传区域中最可能解释GWAS关联的那个转录本。最后得到高置信度风险HERV
以上两步作者合称为rTWAS(retrotranscriptome-wide association study) 补充:
- 为什么先构建遗传调控-基因表达模型,再TWAS:绝大多数GWAS信号在非编码区,不知道具体是哪个基因导致了疾病,这样就构建了
SNP(遗传因素/变异)-转录本(基因表达)-疾病风险的完整链条 - TWAS:这个转录本的预测表达量与疾病风险有没有关联(该位点预测表达量会不会导致更高的疾病风险)。Z值=估计的效应大小/标准误,>0则表达量与风险正相关,<0负相关,绝对值越大则越关联性越强,不同队列得到Z值不能直接比较;P值表示该关联在无真实关联前提下偶然出现的概率(就是统计学里的p值),因为同时检测的转录本数量大,需要做Bonferroni/FDR校正后,才能判断是否“全转录组显著”。|Z|大p小才能说明这个转录本(基因或HERV)的遗传预测表达很可能与疾病风险真实相关
- WGCNA分析:计算任意两个基因或基因-HERV之间的表达相似性矩阵,通过聚类将高度共表达的基因和HERV归为同一模块,统计每个模块中HERV与基因的构成比例,对模块的基因成分进行GO富集分析,推断该模块(及其中HERV)的潜在生物学功能。因为很多HERV的功能尚不明确,通过观察它们与已知基因的共表达关系,可以推测HERV参与的通路或生物学过程
其它补充:
- 转座元件(Transposable Elements, TE):能够在基因组内改变自身位置的DNA序列,可能导致基因表达改变、DNA序列插入/缺失等,进而影响细胞功能。HERV属于TE中的逆转录转座子
- HERV的pol基因:作用是编码逆转录酶(根据RNA模板合成与其互补的DNA,即cDNA),HERV的基因组先通过常规转录产生RNA转录本,在逆转录酶作用下被逆转录为cDNA,通过特定机制(整合酶等)整合到宿主基因组的新位置上。因此HERV属于转座因子——能在宿主基因组内移动,并整合到新的基因组位置;HERV-K LTR作为宿主基因的顺式调控元件,在影响精神分裂症易感基因PRODH的表达
- 含特定HERV特征的基因(genes containing specific HERV features):一个基因与某种类型的HERV序列在基因组上存在明确关系,这些关系具体包括
- 基因内/间插入:HERV位于某个基因的范围内/两个基因之间(同时距离某个基因较近)
- HERV被分为多个超科(superfamily),每个超科下又包含不同的家族或特定序列元素,如果一个基因内部含有来自某个特定家族的HERV序列,就认为该基因含有该特征
- 顺义/反义插入:HERV的转录方向与宿主基因相同/相反,HERV反义转录可能抑制宿主基因
-
主成分分析PCA:从高维数据(如每个样本包含几万个基因的表达值)中找出若干个“方向”,这些方向能最大程度地解释数据中的变化(方差variance)。每个主成分(PC1、PC2、PC3、…)都是数据中一个新的“综合变量”,是原始变量(如各个基因或HERV表达值)的加权组合;PC1表示解释数据中变化最多的方向,PC2是第二多的,依此类推;每个主成分彼此独立,每个主成分解释的百分比,就是这个特征对总体差异的贡献。比如PC1是测序方式、PC2是细胞类型、PC3是疾病状态,就说明测序方式对表达量影响最大,是表达值中最主要的变化来源
在PCA图中,横纵轴通常代表PC1和PC2(因为每个主成分间相互独立,所以PC1⊥PC2),点的分布直观展示根据PC1和PC2进行的分类结果
- 人类参考基因组hgxx的差别:比如hg19是2009年发布的,hg38是2013年发布的,新发布的版本序列更准确,位点注释更全。如果使用的数据(已有的研究成果)是用旧版本做的,就需要把hg19-38相互转换,确保分析基于同一版本
- 协变量(covariates):在基因表达分析、遗传关联分析等统计过程中,可能干扰目标变量(HERV/基因表达水平、疾病风险关联信号)的非核心变量,需通过统计方法(如回归模型调整)排除其影响,以确保分析结果的准确性和可靠性
- 各种
cis-xxx:cis是位点附近上的变异、转录因子、表观等对表达的影响,trans就是远处甚至其他染色体的,更复杂而不易研究-
顺式遗传力(cis-heritability):某个转录本(基因/HERV位点)的表达量,在不同个体之间会有高有低。这些表达差异,有多少能用这个转录本附近的遗传变异(SNP)来解释
只拿附近的一段基因组区域里的SNP,当作预测变量,去预测该转录本的表达。如果由这段“近邻SNP”共同解释的表达方差占比明显高于0(能根据近邻SNP预测出位点表达量),就说这个转录本的表达具有显著的cis遗传成分
- 顺式表达定量性状基因位点(cis-eQTL):位于目标基因/HERV位点附近的遗传变异,且该变异能显著影响该基因/元件的表达水平
-
顺式调控机制/元件(cis-regulatory element):位于目标基因/元件邻近区域的DNA序列(如启动子、增强子、沉默子等),其功能是直接调控该基因/元件的转录/表达过程
顺式调控元件仅影响其物理邻近的基因,反式调控元件(如转录因子基因)可通过表达的蛋白调控远距离基因
- 顺式遗传成分(cis-genetic component):影响某一性状(如基因/HERV表达、疾病风险)的所有顺式遗传因素的总和,包括cis-eQTL、cis-regulatory element的遗传变异等
-
小总结
hERV的研究现状:hERV的结构,对基因组稳定性、基因表达的影响,胚胎发育,免疫系统,人体衰老,致病性
hERV的研究意义:hERV在人类生理过程中的作用和机制;找到参与hERV调节的基因/变异,揭示导致HERV激活的潜在生物学机制;找到哪些HERV会导致疾病(癌症、自身免疫疾病、神经精神疾病、衰老过程),构建根据HERV表达预测疾病风险的模型,对这些HERV进行功能预测以揭示HERV导致疾病的生物学机制,HERV可能作为疾病的生物标记物或治疗靶点
hERV的计算与分析:
-
在基因组注释hERV位置:重复序列检测工具RepeatMasker/RetroTector,重复元件数据库RepBase/DFam、hERV数据库HERVd/EnHERV,人类基因组版本转换工具liftOver主要问题:大部分hERV序列并不完整,许多只剩下单独的LTR序列,或内部携带各种插入/缺失等突变。需要确定哪些序列属于同一个hERV拷贝,可使用REannotate对RepeatMasker输出进行聚合
-
获取各hERV位点的表达量:原始数据很多都是RNA-seq数据,先使用SAMtools将BAM格式转成FASTQ格式(还可此时进行质控的预处理),再使用BLAT/Bowtie2将读段比对到参考基因组,最后使用Telescope/FeatureCounts计算每个hERV位点的表达量主要问题:hERV序列在基因组中有众多相似拷贝,短读长序列很可能比对到多个hERV位置,有的研究中直接使用FeatureCounts丢弃多重比对的读段,有的使用了Telescope这种采用贝叶斯概率模型+EM算法的工具,直接推断出每条读段最可能的来源
-
数据分析:数据预处理(过滤低表达、标准化、校正等),差异表达分析探究哪些hERV位点表达量异常、对hERV影响的基因进行GO富集分析/KEGG富集分析/WGCNA分析解析功能、参与的通路和生物学过程(hERV基因组区段可能缺乏直接功能注释,这时需与其调控的基因一起分析),或者看表达异常的hERV都属于哪个hERV家族、在不同细胞中的富集情况等,癌症研究中构建hERV表达量~疾病风险模型(PCA、生存分析等),或者GWAS/TWAS研究hERV表达和疾病之间的关系,最后找到可作为疾病生物标记物或治疗靶点的hERV
AD与hERV的关系:AD患者出现异常的hERV激活(与Tau蛋白有关),通过调控基因表达等方式影响免疫系统,导致神经炎症和神经退行
- hERV转录异常:HERV-K表达量显著提高,HERV-W、HERV-H、HERV-L等多个家族的转录也呈现上调趋势(尤其是各类env蛋白);一些参与hERV调控的基因呈现高甲基化,部分差异表达的hERV与已知参与AD通路的基因存在共表达相关性,同时这些hERV插入往往靠近这些基因,hERV的异常表达可能通过影响邻近基因表达而影响AD通路(细胞粘附、细胞周期等)
- hERV的激活:AD会导致过度磷酸化的tau蛋白聚集,引发染色质结构的改变,导致曾被沉默的TE/HERV序列露出并重新转录,HERV表达水平与tau病理正相关
- 神经元损伤和炎症:hERV被激活后,其产物可能加剧神经元损伤和炎症,而炎症在刺激ERV表达中起着主要作用,形成恶性循环
- HERV-K(HML-2)的某些转录本(Env蛋白)包含特殊的核苷酸序列模体,能够被TLR8(人类Toll样受体8)直接识别,诱导NF-κB等转录因子,导致TNF-α(肿瘤坏死因子α)等炎性介质的释放,诱导神经元产生氧化应激和凋亡,导致神经元凋亡增加、活化的小神经胶质增多,形成慢性炎症环境;同时,HERV-K转录本具有类病原分子(PAMP)的作用,引发免疫系统攻击
- 激活的hERV的LTR可能通过其强启动子活性,提高邻近基因的表达;同时其所在基因组区域的染色质开放程度升高,可能使转录机制在该区域驻留更长时间,导致相邻的宿主基因也更多地被转录
- 细胞外囊泡(Extracellular Vesicles, EV):衰老和病变的细胞可能将hERV的RNA或蛋白封装进EV中释放到细胞外,它们可以长距离迁移,进入其他神经元或胶质细胞,被称为类逆病毒颗粒(RVLP),这样hERV产物就能作为旁分泌信号,在脑内扩散,同时使hERV成分躲避免疫侦测而不被立即清除,影响更大范围脑组织的炎症和损伤
- 基因组cDNA(gencDNA):来自神经元自身mRNA(特别是APP转录本)的cDNA片段,通过活化的hERV或其他转座子提供的反转录酶作用逆转录并插入基因组,产生拷贝数增多和序列突变,可能促进Aβ过量产生和沉积。
参考文献1:人类内源性逆转录病毒及其在阿尔茨海默病、炎症和衰老发病机制中的假定作用
参考文献2:与阿尔茨海默病相关的位点特异性内源性逆转录病毒表达
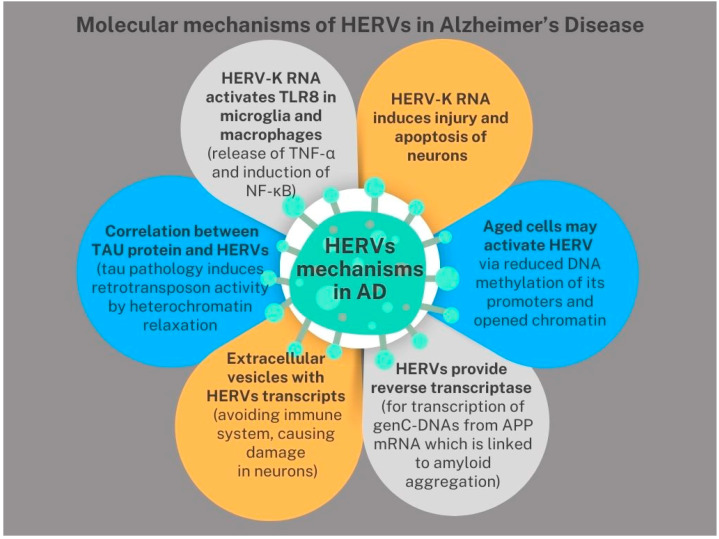
- Tau病理导致异染色质松弛从而激活hERV
- 激活的HERV-K RNA通过TLR8通路引发小胶质细胞和巨噬细胞的炎症反应并直接诱导神经元损伤和凋亡
- 衰老细胞因启动子去甲基化和染色质开放更易激活hERV
- 含hERV转录物的胞外囊泡在神经元间传播损伤信号
- hERV活化提供反转录酶催生来源于APP转录本的基因组cDNA,促进Aβ相关病理
还有什么需要研究的方向、该方向意义是什么、如何解决该方向中可能遇到的问题:
-
AD与hERV激活有强相关性,AD会促进hERV激活,hERV激活又会加速AD的进展。在AD发病前,是否有某种hERV被激活导致AD的发病,还是hERV激活仅仅是AD发病的附加效应(AD发病与hERV激活的因果关系)
可能需要实验验证,比如抑制hERV的表达(通过逆转录酶抑制剂或敲除关键位点),观察是否能减缓AD病变进程
- hERV导致AD相关基因表达改变的具体机制——hERV插入导致基因组序列改变而干扰邻近基因转录/hERV表达产物促进基因表达/染色质结构改变使hERV和邻近基因表达同时改变
- 准确定位是哪些hERV位点影响AD(而不是只在HERV-K、HERV-W等家族层面),验证这些位点的具体作用,位临床治疗/预测提供靶点
- AD患者之间hERV激活程度的差异——患者的个人情况(基因型、年龄性别、病情进展)、环境因素影响(患者是否有病毒感染)
- 临床治疗中评估疗效/特异性/安全性:抗逆转录病毒药物可抑制hERV的活性,减弱其产物对免疫系统的影响,需要进一步验证;或者使用针对HERV-W Env等蛋白的单克隆抗体;或者使用小分子抑制剂阻断hERV激活的免疫通路
- hERV转录本是否能作为AD筛查诊断的指标——需评估其敏感性和特异性,以及与传统AD生物标志物(Aβ、tau蛋白)的关系。但健康人也可能有低水平hERV表达,同时AD患者表达hERV也有差异
- 与单细胞空间转录组结合——探究不同脑区/细胞中的hERV表达是否有差异,hERV的影响是否会在不同区域中传播,不同细胞中的hERV表达差异性是否随时间/病情进展而改变