hERV的计算
other-other2025.10.17-2025.11.7研究进展
文献阅读
Polygenic burden of short tandem repeat expansions promotes risk for Alzheimer’s disease
主题:短串联重复序列(STR, short tandem repeat)与AD发病风险之间的关系
使用的数据:老年AD病例/对照->外周血基因组DNA->无PCR偏好的全基因组测序(WGS)->仅纳入欧洲血统的白人个体和测序深度30×~50×的样本,减少了技术假阳性和混杂因素
研究方法:
- 建立一个多态性STR位点库:在初始的895,826个位点目录上,选取欧洲裔样本,用gangSTR软件筛选出237,197个在群体中长度多态的STR位点,并与已知多态STR集合合并,得到312,731个独特多态STR
- 用ExpansionHunter和gangSTR两款工具对受试者的WGS数据进行STR长度基因分型。为验证短读长测序对STR长度测定的准确性,使用4个样本的长读长测序(PacBio)结果进行对比,综合考虑准确性和灵敏度,后续主要采用ExpansionHunter的基因分型结果
- 关联分析
- 单个STR长度与AD的关联:将每个位点的长度(取每人较长等位基因)作为连续变量,采用logistic回归检验其与AD发病的关联
- 扩增负担分析:将每个位点中超过特定长度阈值的等位基因定义为“扩增”(expansions),比较AD病例与对照中携带该扩增的个体数量差异
- 使用DBSCAN密度聚类算法辨识群体中每个位点长度的异常高值,将这些异常长的等位基因定义为“STR扩增”(识别AD中罕见的长串联扩增),比较病例组与对照组的扩增总负荷差异,评估“高扩增负荷”者的AD患病相对风险
- 功能富集分析:将AD相关的STR扩增位置与染色质状态、组蛋白修饰进行重叠富集检验,并考察AD扩增是否富集于特定转座子元件
主要发现:
- AD病例携带的STR扩增数量显著多于同龄健康对照,STR扩增负荷最高的人群患病风险显著升高(STR扩增累积效应)->累积的STR扩增对疾病风险有实质影响,全基因组多个微效应的重复扩增(Polygenic burden)共同提高AD易感性
- 在单个位点层面,仅发现APOE基因附近的一个STR与AD显著相关,但APOE基因本身就是就是AD的强风险因素之一(此STR与APOE基因连锁),未发现其他STR位点在全基因组水平上达到显著关联(没有一种STR可以独立导致AD的遗传风险)
- AD患者中特有的STR扩增高度富集在活跃的启动子区域(如海马组织的启动子相关染色质区域)。相比身体其他组织,这种富集在活跃启动子/增强子的趋势在大脑组织中最为明显
- AD相关STR扩增显著富集于SVA转座子(神经元中特定基因的调控元件)->STR扩增可能通过影响转座子介导的基因调控来参与AD发生
- AD相关的STR扩增临近的基因高度富集在神经生物学功能(神经元投射形态发生、轴突发育)->AD的STR扩增主要影响神经元结构和信号传递相关的基因
- STR扩增负荷可能成为评估AD遗传风险的一个指标;虽然STR扩增本身很难作为治疗靶点,但其提示的关键通路(例如突触功能、神经发育通路)以及涉及的基因调控机制可能成为新疗法研发的切入点
Structural variation in 1,019 diverse humans based on long-read sequencing
可变数目串联重复序列(VNTR):在基因组中以一段相同或相似的核苷酸序列为重复单位,首尾相连重复的DNA片段,具有高度的遗传多态性和高度重复性。重复单位长度一般在6~70个碱基对之间,重复次数为数次至数百次
- 小卫星DNA的该类序列称为VNTR,微卫星中的则称为STR
- 微卫星DNA:1-6bp组成的短串联重复序列,总长度通常不超过400bp
- 小卫星DNA:15~65bp(6~70bp),总长度通常不超过20kbp
- duplication:复制了一段序列的事件(串联重复拷贝到邻近位置),而VNTR是已存在的串联重复位点上“重复单元拷贝数变化”的事件
- VNTR的插入和缺失:与参考基因组相比,如果某位点的重复数更多就是插入,更少就是缺失
转导(transduction):移动元件在转座时将其源位点邻近的非重复序列一同携带并插入新基因组位置
- 逆转座(retrotransposition):逆转座子L1(Long interspersed nuclear element-1, LINE-1)、SVA(SINE-VNTR-Alu),逆转录时能携带附近的基因组序列一并插入(转导)
- 其中L1有自我复制能力,主要3’端转导;SVA不能自己编码逆转录酶,依赖L1的蛋白,内部有变动很大的VNTR区域,所以大小变化更灵活,3’/5’端转导,多态性插入更多
使用长读长测序对1,019个人的基因组进行了结构变异的全面分析,得到167,291个SV变异位点(107,005个双等位变异位点,其余为多等位变异),其中65,075个为缺失、74,125个为插入,另有25,371个属于复杂类型(参考与替代等位序列均长于1bp);对30万个多等位的VNTR进行了基因分型
- 每个个体基因组中由于SV导致的碱基差异总量约为4.78–9.88Mb,从约50bp的小型SV到数万bp的大型SV均有,长读长测序显著提高了对长度介于几十到几百bp区间SV的识别能力->SV贡献了个人基因组中相当大的变异部分
- VNTR位点的等位基因多样性极高(约一半都是多等位变异)
- 鉴定了一些新的假基因插入,检测到10个HERV-K序列插入以及28个孤立LTR插入事件
- L1与SVA的转导表现出家族与位点特异的偏好
- 分析了变异断点序列的特点,以推断SV形成的分子机制:与DNA的修复机制有关;大量SV的断点被重复元件序列包围,转座元件介导的重组是SV形成的重要机制之一(Alu重复序列、L1序列、LTR元件序列、HERV-K家族)
- 不同人群的SV变异数量和频率存在显著差异,非洲样本贡献更多新SV(有较高的遗传多样性);基于SV的群体结构与地理分层清晰(能通过基于SV基因型的PCA将样本按区域聚类)
- 涉及功能基因组元件(如蛋白编码序列或调控区域)的SV变异其等位基因频率更低->功能相关的SV往往受到更强的纯化选择,符合此类变异可能对生物功能有较大影响的预期
- 少部分SV可能在进化中独立重复地产生(复发突变:同一结构变异在进化过程中多次独立产生)
- 长读长群体SV数据库能够有效剔除患者基因组中与疾病无关的常见或良性SV,缩小遗传诊断的搜索范围,保留绝大部分真正致病的结构变异,推动解析复杂重复区域与疾病的关联(长读长优化了对复杂基因座的分型准确度)。可用于甲基化研究、变异推断与GWAS,并直接提升变异优先级判定与精准医疗
- 未来的鉴定方向:极罕见SV、大倒位、着丝粒/高同源重复区与多等位VNTR;推荐“大队列中等覆盖+子集高深度组装”的策略,需要更可扩展的图算法并与SNP/Indel/STR联合建模
- 大样本中等覆盖+小样本高深度组装:对很多人做中等测序深度,足够发现与分型常见到罕见的多数SV;再选一小部分样本做高深度、多平台、组装级测序把复杂/难区(如着丝粒、超高同源片段重复、超多等位VNTR、大倒位)拼清楚,拿来当模板/锚点改进全体分析
- 极低频SV:样本里出现太少,中等深度下证据弱。大型倒位:拷贝数不变,无法靠覆盖度识别,且断点常在重复序列里,常规比对容易错。着丝粒/高同质性片段重复:重复太长、太像,读段很难唯一定位。多等位VNTR:一个位点好多长度等位,图里会出现多分支,容易对不齐、分型不稳
- 不同的平台优势互补(长度、准确度、跨超长重复)。图算法需要既能扩展到大人群,又能同时建模多种多态性(SNP、Indel、STR/VNTR、SV),减少在重复区的比对歧义与分型错误
微同源序列(microhomology):断点两侧很短的相同小片段,帮末端对齐再连接,一般1–15bp
钝端连接/断点(blunt end/breakpoint):断口没有可配对的突出端或微同源,直接进行连接
移动元件插入(MEI):转座元件(Alu/L1/SVA)插入到新位置
末端双引发(twin priming):L1在同一次插入里,用两处引发点启动逆转录,导致插入序列头端出现小倒位或特殊结构
DNA的修复机制:
- 同源依赖修复(HDR):把一条相似或相同的DNA序列当“模板”,按着模板把断裂处精确补齐。当断点两侧有较长同源序列时,常见到HDR的痕迹
- 非等位同源重组(NAHR):HDR的一种特殊情况,基因组里相似度很高、但位置不同的重复序列错把对方当模板,于是发生错配重组,导致缺失、重复或倒位
- 转座元件介导的重排(TEMR):指由重复元件同源性驱动的一大类重排,常被归到NAHR/HDR范畴里,断点两侧是Alu/L1/LTR等,通过它们的同源性形成SV(比如文章里提到的Alu–Alu、L1–L1、LTR–LTR成对的同源断点)
- 非同源末端连接(NHEJ):不找模板,把多出来的碱基去掉/补齐,直接连接断裂末端。大量断点只有1–15bp的微同源,甚至完全钝端
- 替代末端连接(alt-EJ)/微同源介导修复(MMEJ):类似于NHEJ,会利用很短的“微同源”序列来对齐再粘合,所以断点两侧常见到短短的同源小片段
- 微同源介导的断裂-诱导复制(MMBIR):是DNA复制相关的修复/重排。复制叉因为某些原因而不能继续复制时,聚合酶靠几bp的微同源去别处“搭车”继续复制,容易造成复杂重排、倒位重复等(例如这种机制导致的倒位重复多见1–15bp微同源断点)
一些基础知识补充
hERV的各种分类
Family-level(家族层级):一组具有序列相似性和共同起源的ERV元件,根据其聚合酶基因序列(Pol蛋白序列)的相似性,通常用HERV-X表示,X是它入侵宿主祖先细胞时,所使用的外源性逆转录病毒包膜蛋白的受体类型
- Superfamily(超家族/类别):按逆转录病毒的进化类群划分。
ClassI包括γ和ε逆转录病毒(gamma- and epsilonretroviruses),参与多种生理过程(胚胎发育、免疫通路、各类疾病),包括HERV-W、HERV-H、HERV-FRD、HERV-E、HERV-9等;ClassII包括α、β、δ逆转录病毒和慢病毒(alpha-, beta-, deltaretroviruses, and lentiviruses),最研究深入的家族,与各类疾病密切相关,包括HERV-K等;ClassIII包括spumavirus,最古老,序列残片较多,无表达能力,包括HERV-L等 - Family(家族):一类具有相似序列、共同起源的hERV插入序列,最常用,例如
HERV-K、HERV-W、HERV-H、HERV-L等。家族之间差异很大,有的可能已有表达产物,有的完全失活,包括HERV-L 等 - Subfamily(亚家族):部分比较大的family,进一步分为subfamily。例如HERV-K,可进一步分为
HML-1~HML-10(HML指human MMTV-like,表示它们与小鼠乳腺病毒类似),其中HML-2是其中最年轻、保留编码能力最强的一类,常见于癌症/AD研究中;还有的例如HERV-H按序列相似性分为不同的插入群组(如group 1/group 2),但这类亚分类不太标准化,常用于特定研究
Species-level(种级):某个特定hERV序列在人类基因组中的一个特定插入位点,是进行基因型-表型关联分析的最小单位。最开始一个前病毒整合到了基因组的一个特定位置,之后通过逆转录转座等机制,这个前病毒的“副本”被复制并插入到基因组的其他位置,每一个这样的独立插入单元,都被称为一个Species/Locus
- 例如在某个人类基因组中发现的一个HERV-K元件插入在7q11.23的某个位点,就是一个species-level的元素,基因组上还有很多个HERV-K家族插入在不同位点上,每一个位点都是一个独立单位(Species)
插入基因组后如何影响基因表达
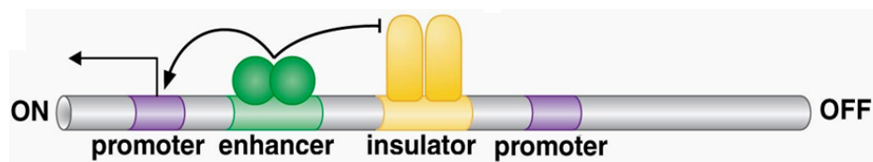
顺式调控:HERV在基因组中作为顺式作用元件,从而影响邻近基因的转录——HERV的LTR具有启动子/增强子/绝缘子的功能,含有转录起始位点及转录终止(polyA)信号
-
作为基因的替代启动子,启动该基因的转录
启动子(promoter):RNA聚合酶开始转录的结合位点
-
作为增强子提高邻近基因转录,依赖LTR序列中存在的转录因子结合位点(TFBS)
增强子(Enhancer):与特异转录因子结合的DNA短区,在任何方向、上游或下游调控转录
-
作为绝缘子或包含转录终止信号,影响基因表达边界,LTR序列两端的U5和U3区可携带终止转录的polyA信号,可导致上游转录在此提前终止
绝缘子(Insulators):位于增强子和启动子之间的调控序列,与绝缘子结合蛋白结合,确保某个增强子只影响特定的启动子,阻隔远端增强子对基因的不恰当作用
反式调控:通过其转录产物(蛋白或RNA)对远距离的基因进行调控
- HERV蛋白质:直接充当新的细胞因子或膜蛋白,影响细胞通讯,或者通过干扰宿主转录因子、受体通路等间接改变基因表达。例如HERV-K(HML-2)家族编码的Rec和Np9蛋白可以物理结合一种转录抑制因子(PLZF,一种肿瘤抑制因子),从而解除PLZF对其靶基因的转录抑制,导致致癌基因过度表达
- HERV非编码RNA:HERV产生的长非编码RNA或反转录病毒粒子RNA也能在细胞内迁移,并影响远端基因表达。例如长非编码RNA可结合调节转录因子复合物,从而改变相应基因表达;HERV的RNA可能被包装进外泌体或病毒样颗粒,在细胞间传递影响邻近细胞的基因表达模式(比如肿瘤微环境中引起周围免疫细胞活化)
表观遗传:人体通过多种DNA甲基化和组蛋白修饰机制,使大部分HERV元件被高度DNA甲基化覆盖,并缠绕在异染色质中,从而转录沉默
- 当一个HERV插入到某个基因/基因调控区旁边时,可能通过DNA甲基化/组蛋白修饰/异染色质传播影响邻近的基因。例如当插入序列高度甲基化并为异染色质形态时,可能将沉默效应带到邻近基因,导致该基因转录受抑;某些情况下,HERV插入的存在会引发局部去甲基化或异染色质边界改变,使邻近基因意外激活或表达失调
- HERV插入序列的转录因子结合位点和结构蛋白结合位点也可能发生表观遗传修饰变化,影响相应转录因子的结合和染色质构象,进而改变基因表达
干扰基因转录:
- 如果HERV以与宿主基因相反的方向插入,那么HERV的LTR(作为启动子)可能驱动产生一条与宿主基因mRNA互补的反义RNA,并与宿主mRNA形成双链RNA,从而阻碍mRNA的翻译
- 与宿主基因竞争转录因子结合位点/启动子和增强子
- 可能引入新的剪接供体或受体位点,扰乱原有剪接信号,导致产生异常可变剪接产物
- HERV序列中存在潜在的剪接供体/受体位点,插入基因内含子后,其LTR或内部序列可以充当新的外显子
- 破坏原有剪接信号或引入竞争性剪接信号,从而改变宿主基因的剪接模式,比如使本来被隐蔽的剪接位点显现,替代原本的剪接位点
- 与邻近基因拼接成嵌合转录本/截短转录本:HERV作为新启动子或polyA信号,使转录提前从HERV序列开始或在HERV处终止,或者插入在外显子内,导致部分HERV序列作为插入外显子拼接到正常mRNA中
其它:
- 双链RNA可能会被Dicer酶识别而产生siRNA,沉默具有相同序列的转录本
- 部分HERV序列被识别成miRNA基因,产生成熟miRNA,降解或抑制特定宿主mRNA实现对细胞通路的调控
- 部分HERV转录本带有miRNA结合位点,通过竞争抑制,上调那些miRNA原本抑制的基因
- 通过“类病毒效应”在宿主抗感染通路中影响相关基因表达
- 部分HERV序列带有结构蛋白结合位点,重塑更大范围的染色质三维结构:形成新染色质边界,影响基因调控;改变染色质环的拓扑结构,影响异染色质传播
TE/剪接相关
基因的5’端是贴近TSS/启动子的一侧;3’端是贴近TES/加尾的一侧,方向相对该基因的转录方向
- TSS(Transcription Start Site):转录起始位点,一般位于启动子区域附近
- TES(Transcription End Site):转录终止位点,通常与剪切-加尾位点(cleavage&polyadenylation site, PAS)重合,mRNA在此被切断并加上polyA尾
转录生成前体mRNA(pre-mRNA),里面有外显子和内含子,剪接体识别5’剪接位点(内含子5’端的外显子-内含子边界)和3’剪接位点(内含子3’端的外显子-内含子边界),在这两个位点切割,将内含子切掉,外显子拼接在一起
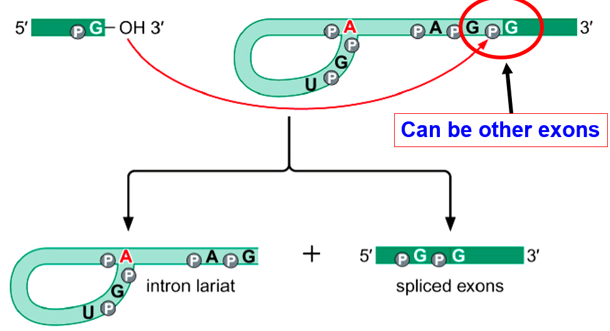
5’-TSS–启动子–外显子序列1–5’剪接位点–内含子–3’剪接位点–外显子序列2–终止子–TES-3’
许多TE(比如LTR型的hERV家族)自身携带启动子/增强子、可作为外显子的一部分,并且内部或邻近序列常含剪接位点或PAS信号,当TE插入到基因附近或基因内,并与基因同向时,可能发生三类“结构重写”
- TE携带启动子和TSS,这个TSS转录出的第一个外显子来自TE序列,相当于把原基因的外显子序列1延长了一段,出现替代第一外显子(Alternative First Exon, AFE),形成TE-基因的嵌合转录本(TE-initiated/LTR-initiated transcript, LIT)
- TE-介导的内部外显子化(internal exonization):TE插在内含子里,其中含有5’/3’剪接位点,剪接体把TE的一段当作外显子夹在两个基因外显子之间
- TE内部含有TES/PAS信号,插入到原基因的TES前,作为新的加尾终止位点,导致转录更早终止,且剪接时外显子序列2含有一段来自TE的序列,出现替代最后外显子(Alternative Last Exon, ALE),形成TE-基因的嵌合转录本
- 若TE与基因反向,依然可能产生反义转录并影响剪接/干扰
计算流程
ENCODE数据+bowtie2比对+Telescope计数
序列下载与比对
配置环境:
conda create -n telescope -c conda-forge -c bioconda \
python=3.10 telescope bowtie2 samtools sra-tools parallel-fastq-dump \
flexbar bedtools entrez-direct pigz -y
conda activate telescope
下载ENCODE数据:以SRP014320中的H1-hESC细胞系为例
# 列出 SRP014320 的全部测序结果,并保存 CSV
esearch -db sra -query SRP014320 | efetch -format runinfo > SRP014320_runinfo.csv
# 以 H1-hESC 为例筛选
grep -i 'H1' SRP014320_runinfo.csv > H1_runinfo.csv
# 提取 SRR 列
awk -F, 'NR>1 {print $1}' H1_runinfo.csv > SRR_H1.txt
为了节省磁盘空间,只取SRR_H1.txt的前两个数据SRR521515和SRR521514
# 并行下载并直接生成FASTQ
parallel-fastq-dump --sra-id $(head -n1 SRR_H1.txt) --threads 8 --split-files --outdir fastq
# 质控/剪接
flexbar -r fastq/$(head -n1 SRR_H1.txt)_1.fastq -p fastq/$(head -n1 SRR_H1.txt)_2.fastq \
-t fastq/$(head -n1 SRR_H1.txt)_trim --adapter-trim-end RIGHT --min-read-length 30
- 下载的数据格式为
sampleX_1.fastq+sampleX_2.fastq,说明是双端测序结果(paired-end sequencing),经比对后输出到一个BAM中,BAM中每条记录带有配对标记 - 最后生成一个fastq文件夹,主要包含4个文件,格式为
SRR521514_trim_1.fastq和SRR521514_trim_2.fastq
注:这里$(head -n1 SRR_H1.txt)是取txt里的第一行样本,没有设置循环,需要手动执行两次(下面类似)
下载人类参考基因组hg38:
mkdir refs && cd refs
wget https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/latest/hg38.fa.gz
gunzip hg38.fa.gz
bowtie2-build hg38.fa hg38
cd ..
- 生成的refs文件夹即为hg38参考基因组
下载HERV注释:直接用作者提供的HERV_rmsk.hg38.v2/genes.gtf
# 需要先下载git lfs
sudo apt-get install git-lfs
git lfs install
git clone https://github.com/mlbendall/telescope_annotation_db
# 根据人类参考基因组版本选择注释文件路径
ANN=./telescope_annotation_db/builds/HERV_rmsk.hg38.v2/genes.gtf
注:后来仔细看了下GitHub仓库,好像应该用同路径下的transcripts.gtf作telescope的注释
比对到基因组:使用论文参数运行时发现时间非常长,于是调整参数
SRR="SRR521515"
bowtie2 -x refs/hg38 -1 fastq/${SRR}_trim_1.fastq -2 fastq/${SRR}_trim_2.fastq \
# --very-sensitive-local -k 100 --score-min L,0,1.6 -p 8 \ # 论文参数
--sensitive-local -k 20 --score-min L,0,1.6 -p 8 \ # 为节省时间调整后参数
2> ${SRR}.bowtie2.log | samtools view -bS - | samtools sort -o ${SRR}.sorted.bam
SRR="SRR521514"
bowtie2 -x refs/hg38 -1 fastq/${SRR}_trim_1.fastq -2 fastq/${SRR}_trim_2.fastq \
# --very-sensitive-local -k 100 --score-min L,0,1.6 -p 8 \ # 论文参数
--sensitive-local -k 20 --score-min L,0,1.6 -p 8 \ # 为节省时间调整后参数
2> ${SRR}.bowtie2.log | samtools view -bS - | samtools sort -o ${SRR}.sorted.bam
- 生成
SRR521515.sorted.bam和SRR521514.sorted.bam
观察这两个样本是否为同一个H1样本的技术重复:
# 分别拉取 runinfo(CSV),看关键列是否一致
esearch -db sra -query SRR521514 | efetch -format runinfo > SRR521514.csv
esearch -db sra -query SRR521515 | efetch -format runinfo > SRR521515.csv
查询SRR521514.csv和SRR521515.csv
- 同一Experiment且同一BioSample:同一文库被多次上机,是技术重复,可合并作为一个样本来运行后续步骤
- Experiment不同但BioSample相同:同一生物样本做了不同文库;通常也按技术重复处理,但合并前要确认LibraryStrategy/Layout/读长/建库是否一致;也可以分别运行,最后合并基因表达量
- BioSample不同:不是同一样本(一般是生物学重复),不要合并
这里发现Experiment和BioSample都相同,因此运行下面的合并命令
samtools merge H1_sample.merged.bam SRR521515.sorted.bam SRR521514.sorted.bam -@ 8
- 生成
merge H1_sample.merged.bam——整合后的bam文件
telescope:需要输入的BAM文件不能是坐标排序,必须samtools sort -n或samtools collate
samtools sort -o H1_sample.merged.sorted.bam H1_sample.merged.bam -@ 8
samtools index H1_sample.merged.sorted.bam
samtools collate -@ 8 -o H1_sample.collate.bam H1_sample.merged.sorted.bam
- 最后生成
H1_sample.collate.bam——排序后的比对结果
先检查telescope是否能正常运行
eval $(telescope test)
- 正常会生成一个
telescope-telescope_report.tsv,我这里报错AttributeError: module 'numpy' has no attribute 'int'.,查询GitHub issue后发现是作者的源码有问题。把/home/userName/miniconda3/envs/telescope_env/lib/python3.10/site-packages/telescope/utils/model.py里面的np.int改成int即可
mkdir telescope_out
telescope assign H1_sample.collate.bam ${ANN} \
--outdir telescope_out --exp_tag H1_sample \
--theta_prior 200000 --max_iter 200
步骤详解
ENCODE数据:可以在ENCODE官网下载,也常常同步到NCBI SRA数据库
SRP:项目编号(例如”ENCODE Caltech RNA-seq”)SRX:项目中某次实验的编号(标明建库类型、读长/平台等)SRS:实验中某个样本的编号(例如”细胞系/细胞类型H1-hESC”)SRR:对样本测序时某次上机/测序运行的编号,同一个SRX/SRS可能有多个SRR- 技术重复:同一文库多次上机,可以合并BAM文件,当作同一个样本
- 生物学重复:不同个体/细胞培养批次的样本,分析时要作为独立样本,分别定量
bowtie2比对:
- 输入:将成对或单端的FASTQ(原始reads),Bowtie2索引(例如这里的人类参考基因组hg38)
- 目的:为每条read找出在基因组中的匹配位置
- 对于hERV/TE,因为基因组上存在很多重复序列,所以很多reads有多个同样好的位置,因此用
-k 100保留多重匹配(最多保留100条匹配记录) --very-sensitive-local/--sensitive-local:更宽松、允许局部匹配,能识别截断或带错配的TE片段--score-min L,0,1.6:放宽打分阈值,让低错配但多拷贝的reads不被扔掉
- 对于hERV/TE,因为基因组上存在很多重复序列,所以很多reads有多个同样好的位置,因此用
- 输出:SAM文件(纯文本)
samtools view -bS -:将SAM转换成BAM(二进制文件,省空间、读写快)samtools sort:按坐标(染色体/位置)排序,是很多软件常用的格式(能建索引、能快速随机访问、便于可视化)samtools collate/samtools sort -n:按read名称排序,不能建索引samtools index:给按坐标排序的BAM文件建索引,生成.bai文件,方便后续查看、统计samtools merge:把多次上机的技术重复合并成一个BAM,避免后面重复计算
Telescope计数:
- 输入:按read名称聚集的BAM文件、hERV注释GTF
- 目的:把多重比对的读段匹配到最可能的单个hERV位点,如果置信度低则丢弃(不会同时给多个位点加分)
- 把唯一比对的reads直接记入对应位点,多重比对的reads用EM/贝叶斯模型在候选位点间重分配,直到收敛
- 因为hERV/TE多拷贝/高相似度的特性,同一条read会有多条比对记录。而Telescope在重分配时需要一次性拿到这条read的所有候选位置,要求BAM里同一read的多条匹配记录挨在一起,而坐标排序会把这几条记录分散到整条染色体,telescope无法正确聚合
--theta_prior:给“歧义分配”一个惩罚型先验,防止把噪声硬塞进错误位点--max_iter:迭代上限--reassign_mode average:一个读段在并列最佳的几个位点间均分到计数中
- 输出:每个hERV位点(Species-level)的表达数量
- 优势:如果只计数唯一比对,可能会丢掉大量多重比对的读段。而telescope同时利用唯一与多重信息,用EM模型将读段按全局一致性重分配,更有利于比对species-level——能把同一家族不同基因座的表达区分开
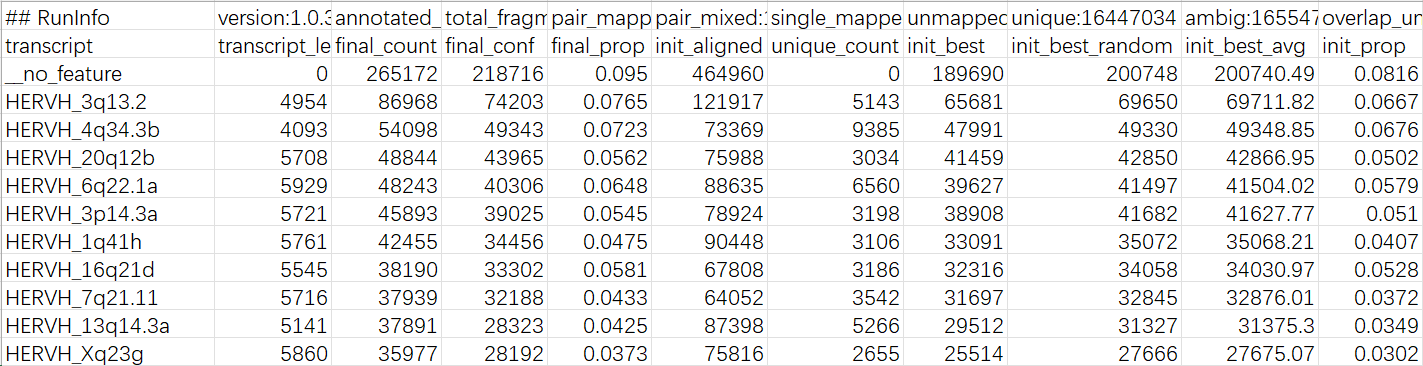
Telescope结果表格:
transcript:位点/转录单元ID(来自注释GTF文件的locus等属性)final_count:最终分配给该位点的片段数(常用于后续进行差异表达等分析)final_conf:高置信(后验≥阈值)的分配片段数final_prop:该位点的最终比例参数π(样本内归一化的相对丰度)unique_count:唯一比对到该位点的片段数(无需重分配的片段数)init_aligned:初始化时“对这个位点有命中”的片段数(多重比对被重复计入时的情况)init_best/init_best_random/init_best_avg/init_prop:不同初始策略下的计数或π,用来对比“EM之前”的起点与“EM之后”(final_*)的差异
featureCounts+TEtranscripts
序列下载和比对、构建gtf文件
为了作差异分析,需要再找一个不同细胞系的数据:根据前面telescope的方法,从SRP014320这个项目里选”K562”这个细胞系里的SRR521457和SRR521464两个技术重复(LibraryLayout为paired的,碱基/spot数接近)
生成BAM比对文件(步骤同ERVmap):
threads=10
# 下载序列
mkdir fastq && cd fastq
echo -e "SRR521514\nSRR521515" >> cell1.srr
echo -e "SRR521457\nSRR521464" >> cell2.srr
for SRR in $(cat cell1.srr cell2.srr); do
parallel-fastq-dump --sra-id ${SRR} --threads ${threads} --split-files
done
# 去接头、质控
cat > adapters.fa <<EOF
>TruSeq3-PE-Read1
AGATCGGAAGAGCACACGTCTGAACTCCAGTCA
>TruSeq3-PE-Read2
AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT
EOF
for SRR in $(cat cell1.srr cell2.srr); do
flexbar -r ${SRR}_1.fastq -p ${SRR}_2.fastq \
-t ${SRR}_trim \
-a adapters.fa \
-m 30 -n ${threads} \
2> ${SRR}_flexbar.log
done
cd ../
# 构建bowtie2参考基因组
mkdir refs && cd refs
sudo apt-get install aria2
aria2c -x 16 -s 16 https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/latest/hg38.fa.gz
gunzip hg38.fa.gz
bowtie2-build hg38.fa hg38
cd ../
# 比对到基因组(推荐使用--very-sensitive -k 100,这里为了节省时间空间只用-k 20)
for SRR in $(cat fastq/cell1.srr fastq/cell2.srr); do
bowtie2 -x refs/hg38 -1 fastq/${SRR}_trim_1.fastq -2 fastq/${SRR}_trim_2.fastq \
-k 20 \
--no-mixed --no-discordant \
-p ${threads} \
2> fastq/${SRR}.bowtie2.log \
| samtools view -bS - \
| samtools sort -o fastq/${SRR}.sorted.bam
samtools index fastq/${SRR}.sorted.bam
done
# 合并技术重复
samtools merge -@ ${threads} fastq/H1.merged.bam $(awk '{print "fastq/"$1".sorted.bam"}' fastq/cell1.srr)
samtools sort -@ ${threads} -o fastq/H1.merged.sorted.bam fastq/H1.merged.bam
samtools index fastq/H1.merged.sorted.bam
samtools merge -@ ${threads} fastq/K562.merged.bam $(awk '{print "fastq/"$1".sorted.bam"}' fastq/cell2.srr)
samtools sort -@ ${threads} -o fastq/K562.merged.sorted.bam fastq/K562.merged.bam
samtools index fastq/K562.merged.sorted.bam
注:这里也可以用samtools collate排序,这样后面TEtranscripts时就不用--sortByPos参数,节省运行时间和空间
bowtie2的比对参数设置:
-k 100:因为后续TEtranscripts的--mode multi和featureCounts的-M -O --fraction都会计入多重比对,所以在生成BAM是就要保留multi-mappers--sensitive/--very-sensitive(-local):没有硬性要求,保持前后分析一致即可,是“更容易收录边界/错配的读段——稍快/更严格”的问题--no-mixed(丢弃“只配对的一端命中”的read-pair)/--no-discordant(丢弃“配对相互位置关系异常”的命中):TEtranscripts官方文档给出的配对端约束建议,让BAM里里只保留“成对且合理”的配对端比对
生成hERV的gtf注释:两种方法,稍后比较下结果
mkdir gtfs && cd gtfs
wget -c https://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/rmsk.txt.gz
# Gene GTF(GENCODE GRCh38,CHR版)——TEtranscripts使用
aria2c -x 16 -s 16 https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_38/gencode.v38.annotation.gtf.gz
gunzip -f gencode.v38.annotation.gtf.gz
# TE GTF(官方提供的GRCh38 GENCODE匹配版)——TEtranscripts和featureCounts使用
# aria2c -x 16 -s 16 https://labshare.cshl.edu/shares/mhammelllab/www-data/TEtranscripts/TE_GTF/GRCh38_GENCODE_rmsk_TE.gtf.gz
# gunzip -f GRCh38_GENCODE_rmsk_TE.gtf
本来是按上面的方法下载,但这个url现已404,使用自制转座子GTF
zcat rmsk.txt.gz | awk 'BEGIN{OFS="\t"}{
chr=$6; start=$7+1; end=$8; strand=$10; repName=$11; repClass=$12; repFamily=$13;
fam=repName; gsub("[-_/].*","",fam);
if(repClass=="LTR" && repName ~ /^HERV/){
tid=repName"_"chr"_"start"_"end;
print chr,"hg38_rmsk","exon",start,end,".",strand,".",
"gene_id \""fam"\"; transcript_id \""tid"\"; family_id \""repFamily"\"; class_id \""repClass"\";";
}
}' > hg38_hERV_family_1.gtf
zcat rmsk.txt.gz | awk 'BEGIN{OFS="\t"}
function herv_family(repName, repFamily, fam){
# 1) 先用 repName 的显式 HERV 名
if (repName ~ /^HERVK/) return "HERVK"
if (repName ~ /^HERVH/) return "HERVH"
if (repName ~ /^HERVW|^HERV17/) return "HERVW"
if (repName ~ /^HERV9/) return "HERV9"
# 2) 再把常见 LTR 名映射到对应 HERV family
if (repName ~ /^LTR5(_Hs|A|B)?/) return "HERVK" # LTR5/LTR5_Hs 属于 HERVK(HML-2)
if (repName ~ /^LTR7/) return "HERVH" # LTR7 系列属于 HERVH
if (repName ~ /^LTR17/) return "HERVW" # LTR17 系列属于 HERVW
if (repName ~ /^LTR12/) return "HERV9" # LTR12 系列多归于 HERV9
# 3) 其它情况:不归类(返回空字串 -> 这条记录将被丢弃)
return ""
}
{
# rmsk.txt.gz 的字段:... 6:chrom, 7:genoStart, 8:genoEnd, 10:strand, 11:repName, 12:repClass, 13:repFamily
chr=$6; start=$7+1; end=$8; strand=$10; repName=$11; repClass=$12; repFamily=$13;
# 只保留 LTR 类
if (repClass!="LTR") next;
fam=herv_family(repName, repFamily);
if (fam=="") next; # 未能映射到 HERV 家族的忽略
# TEtranscripts 约定:feature 用 exon;基因/家族聚合用 gene_id;每个拷贝的唯一ID 用 transcript_id
# family_id 用更细的子家族(这里放 repName),class_id 放 LTR
tid=repName"_"chr"_"start"_"end;
print chr,"rmsk","exon",start,end,".",strand,".",
"gene_id \""fam"\"; transcript_id \""tid"\"; class_id \"LTR\"; family_id \""repName"\";";
}' > hg38_hERV_family_2.gtf
# 在基因组层面:统计family拷贝数&覆盖碱基
zcat rmsk.txt.gz | awk 'BEGIN{OFS="\t"}{
chr=$6; start=$7; end=$8; repName=$11; repClass=$12;
if(repClass=="LTR" && repName ~ /^HERV/){
fam=repName; gsub("[-_/].*","",fam);
len=end-start; CNT[fam]++; BP[fam]+=len;
}
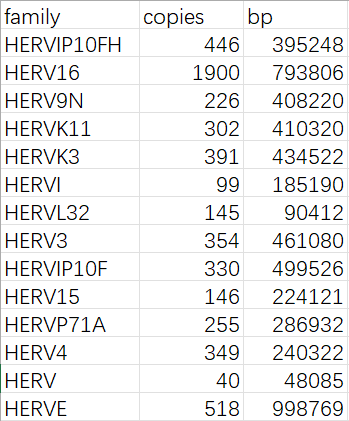
} END {print "family","copies","bp";
for(f in CNT) print f, CNT[f], BP[f];}' > hERV_family_copy_bp.tsv
cd ../
从UCSC的RepeatMasker表rmsk.txt.gz里筛出hERV(repClass==LTR且repName以HERV开头),并构造一份GTF注释,把family写进gene_id;第二个gtf与第一个的区别是把LTR(repName没有以HERV开头但也属于hERV的)也映射到了对应HERV family。最终得到的hg38_hERV_family_2.gtf约是_1.gtf大小的两倍
- 补充——是否可以用telescope的gtf:featureCounts可以,TEtranscripts理论可行,但官方的设计目的是统计家族级别(基因级别),而telescope的gtf是位点级的注释,而且telescope的gtf里没有classid和familyid,需要将geneid改成family、添加classid为”LTR”、添加familyid为repName/intModel,并将feature改成exon
hERV_family_copy_bp.tsv:统计每个family的条目数copies与覆盖碱基bp
单样本分析——使用featureCounts:以H1细胞系的bam为例
conda install -c bioconda subread # 安装featureCounts
mkdir RM_FC && cd RM_FC
featureCounts -a ../gtfs/hg38_hERV_family_1.gtf -t exon -g gene_id \
-o H1_gtf1_counts_familyLevel.txt \
-M -O --fraction -T ${threads} -p -B -C \
../fastq/H1.merged.sorted.bam
featureCounts -a ../gtfs/hg38_hERV_family_2.gtf -t exon -g gene_id \
-o H1_gtf2_counts_familyLevel.txt \
-M -O --fraction -T ${threads} -p -B -C \
../fastq/H1.merged.sorted.bam
cd ../
结果:
-
xxx.txt:样本在family-level上每个hERV家族的所在位点(染色体位置)和表达量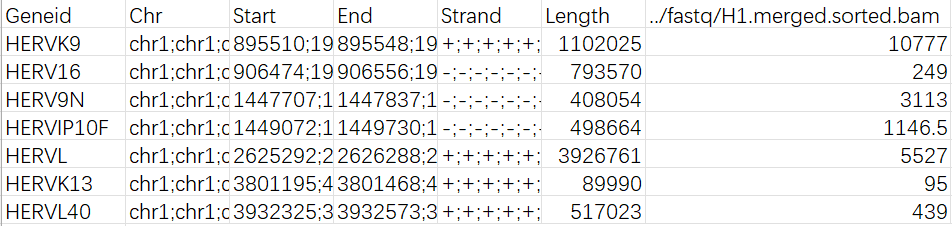
-
xxx.txt.summary:计数的统计结果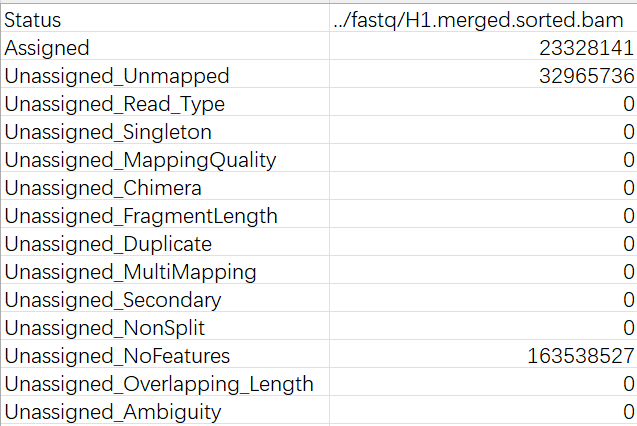
# 安装TEtranscripts
cd /home/lwl
git clone https://github.com/mhammell-laboratory/TEtranscripts
cd TEtranscripts
python setup.py install
TEtranscripts --version # 输出:TEtranscripts 2.2.3
# 安装DESeq2,参考文章:https://zhuanlan.zhihu.com/p/164886255
conda create -n tetranscripts
conda activate tetranscripts
conda install r=3.6
conda install r-xml
conda install -c r r-data.table=1.12.2
conda install -c bioconda bioconductor-deseq2=1.24.0
R # 打开R环境
install.packages("BiocManager")
BiocManager::install("GenomeInfoDb") # 不设置镜像,需要挂梯子(国内镜像站没有旧版本BiocManager)
library("DESeq2") # 检查是否安装成功
# CTRL+Z退出R环境
单样本分析——使用TEcount:以H1细胞系的bam为例
mkdir TE && cd TE
mkdir H1_gtf1_TEcount
TEcount --sortByPos --format BAM --mode multi \
-b ../fastq/H1.merged.sorted.bam \
--GTF ../gtfs/gencode.v38.annotation.gtf \
--TE ../gtfs/hg38_hERV_family_1.gtf \
--stranded no \
--outdir H1_gtf1_TEcount \
2> H1_gtf2_TEcount.log
mkdir H1_gtf1_TEcount
TEcount --sortByPos --format BAM --mode multi \
-b ../fastq/H1.merged.sorted.bam \
--GTF ../gtfs/gencode.v38.annotation.gtf \
--TE ../gtfs/hg38_hERV_family_2.gtf \
--stranded no \
--outdir H1_gtf2_TEcount \
2> H1_gtf2_TEcount.log
TEtranscripts_out.cntTable:每个基因的表达量
多样本差异表达分析——使用TEtranscripts:
mkdir H1_K562_gtf1
TEtranscripts --sortByPos --format BAM --mode multi \
-t $(awk '{printf "../fastq/%s.sorted.bam ",$1}' ../fastq/cell1.srr) \
-c $(awk '{printf "../fastq/%s.sorted.bam ",$1}' ../fastq/cell2.srr) \
--GTF ../gtfs/gencode.v38.annotation.gtf \
--TE ../gtfs/hg38_hERV_family_1.gtf \
--stranded no \
--project H1_K562_gtf1 --outdir H1_K562_gtf1 \
2> H1_K562_gtf1.log
mkdir H1_K562_gtf2
TEtranscripts --sortByPos --format BAM --mode multi \
-t $(awk '{printf "../fastq/%s.sorted.bam ",$1}' ../fastq/cell1.srr) \
-c $(awk '{printf "../fastq/%s.sorted.bam ",$1}' ../fastq/cell2.srr) \
--GTF ../gtfs/gencode.v38.annotation.gtf \
--TE ../gtfs/hg38_hERV_family_2.gtf \
--stranded no \
--project H1_K562_gtf2 --outdir H1_K562_gtf2 \
2> H1_K562_gtf2.log
结果:
-
xxx.cntTable:各样本每个基因/HERV的表达量(HERV相关的结果都显示在表的最后一块,下同)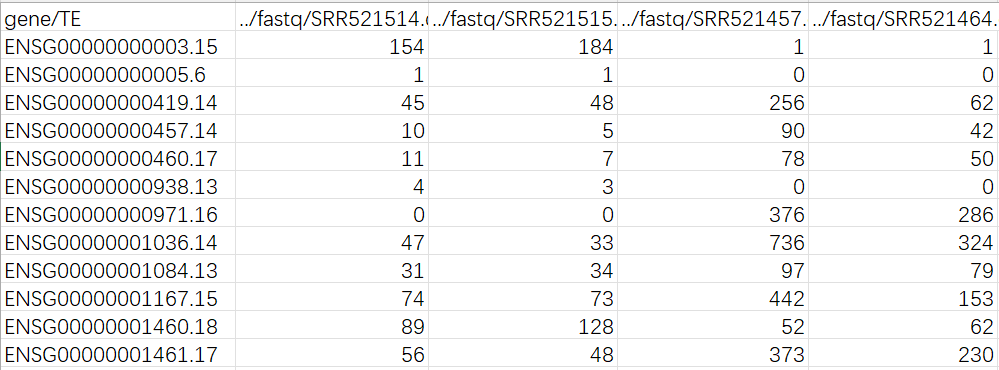
-
xxx_gene_TE_analysis.txt:每个基因的差异表达分析结果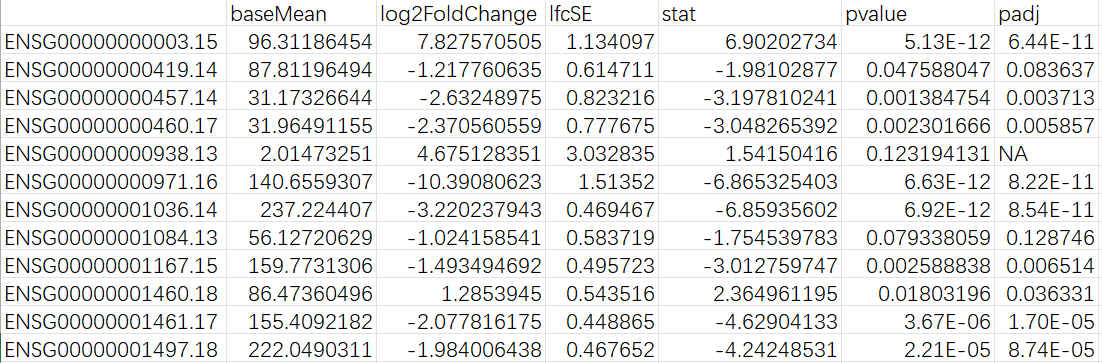
-
xxx_sigdiff_gene_TE.txt:根据指定阈值筛选得到的差异基因(padj<0.05,|log2FC|>0)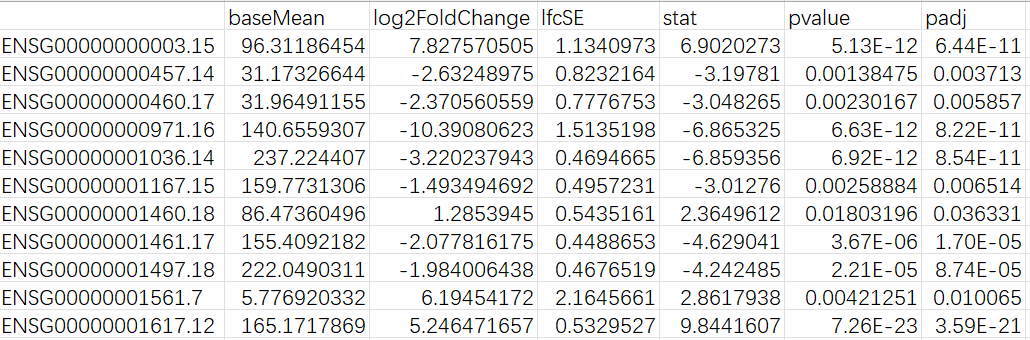
baseMean:对所有样本做归一化后,该基因的平均表达量log2FoldChange:处理组-对照组的log2倍数变化lfcSE:log2FC的标准误(SE)stat:检验统计量(Wald检验时约为log2FC/SE)pvalue/padj:原始/FDR校正后p值
其它问题与后续分析
在下载fastq数据时遇到的问题:
- 发现有的样本(SRRxxx)非常大(10多个G),有的只有几个G:测序深度不同导致reads数不同,在SRA的RunInfo里的spots/avgLength字段标识了这一信息。除此之外,读长越长fastq越大,有的fastq文件可能已经做过过滤/裁剪
-
H1组的样本与K562组的样本大小不一样,是否会对最终结果有影响:通常深度越高效果越好,低深度的样本可能会漏掉低表达的基因,影响差异分析结果。DESeq2等差异分析软件可以通过归一化等方式消除测序深度的影响
注意:归一化能消除“测序量不同”这个偏差,但不能替代足够的测序深度,要确保两组样本的深度没有太大差别(比如一组2×5000万reads,另一组2×500万就不行,深度低的那组检测力会明显下降,方差估计更不稳)
如果不确定,可以直接用原始计数进行DESeq2,不要CPM/TPM等归一化处理后再做差异
下载的同一细胞系的不同样本是否合并:可以合并,但TEtranscripts可以分析这种一组多样本的情况,而且这样的效果更好,所以就保留合并前的原始BAM文件,在TEtranscripts分析时进行“2 vs 2”的差异分析
构建hERV的gtf:
repClass=="LTR":只取LTR类的转座子,在rmsk表里,每条重复都有三元标签:repClass/repFamily/repName。其中repClass是大类(如LTR/LINE/SINE/DNA/Simple_repeat等),HERV属于LTR类反转座子repName~/^HERV/:选出以HERV开头的LTR,因为HERV的条目本来就标成LTR类,所以有无repClass=="LTR"影响不大- hERV的env/gag/pol等基因在哪:rmsk不会按基因粒度注释,而是把前病毒结构分成
两端(LTR)与内部区(internal),内部区常以-int结尾,如HERVK-int,HERVH-int)。env/gag/pol都包含在这个内部区里
TEtranscripts需要的gtf注释有特殊要求:
- 使用
exon作为feature - 属性里要包含
gene_id、transcript_id,并建议加上class_id(如LTR)与family_id(更细的子家族/亚家族名)
小总结:三种计算hERV表达量的工具Telescope、featureCounts、TEtranscripts的特点以及优缺点
- Telescope:针对位点级(Species-level)hERV定量,使用EM/贝叶斯方法分析多重比对
- featureCounts:通用计数器,family-level快速计数,多重比对用分数计数是近似(对高相似家族可能有偏差)
- TEtranscripts:family/Subfamily-level快速计数计数与差异表达,使用EM分析多重比对
因为使用的gtf格式不同,gtf1最后生成的结果基因id格式为HERVIP10FH:ERV1:LTR,gtf2为HERVK:HERVK9-int:LTR,画图时gtf1就直接取HERVIP10FH作为每个点的标注,gtf2按HERVK作图例、HERVK9-int作为标注
HERVIP10FH是subfamily,对应RepeatMasker注释里的repNameERVL是family,对应RepeatMasker注释里的repFamilyLTR是classLTR7这种是生成gtf时,将LTR名归类到HERVx
在gtf1的结果中,既有HERVK的family,又有HERVIP10F这种subfamily:有些repName(如HERVIP10F)没有映射到HERVK/HERVH/HERVW/HERV9,就被原样保留了
在telescope的统计结果中,得到的结果虽然是位点级别,能不能直接按家族合并,生成家族级别的统计结果:理论可行,直接合并同一家族下的所有位点的final_count,将原始计数矩阵直接用DESeq2做差异分析。问题在于要准确定义“家族”的归并规则(比如是否把LTR和-int系列也并入相应家族)
普通基因的点用蓝色标注,HERV的用彩色标注,阈值padj>0.05、|log2FC|>1,按padj排序前15的点标注了名称
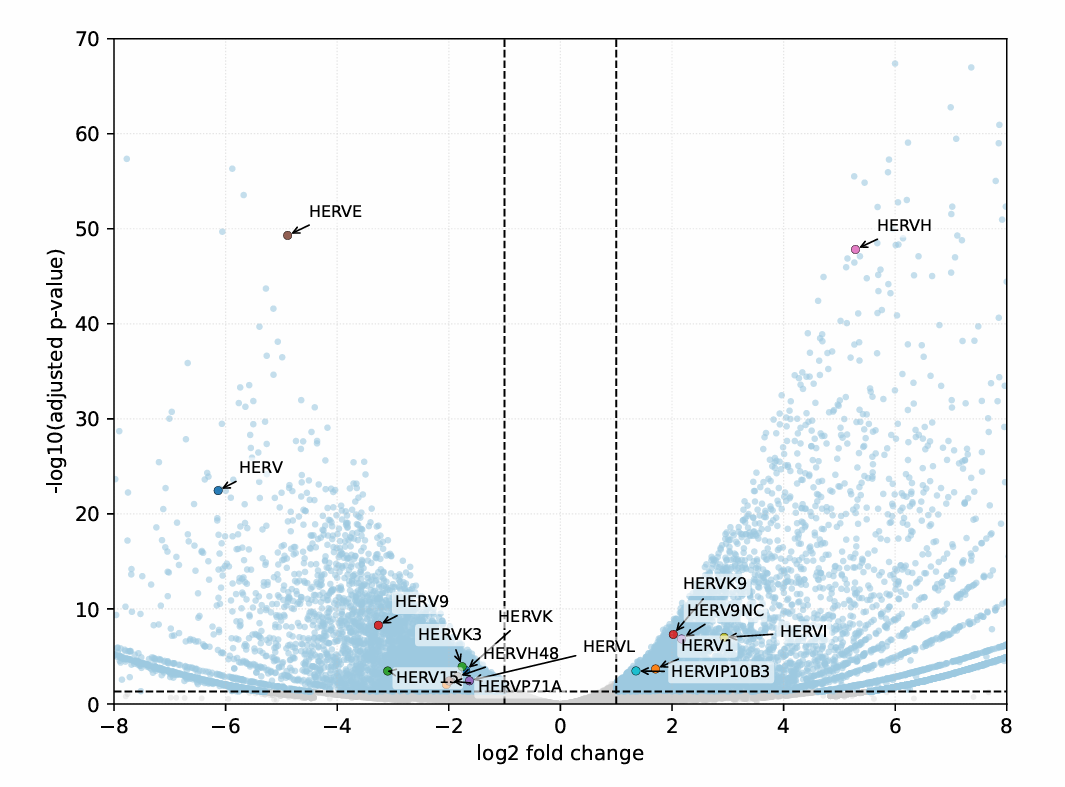
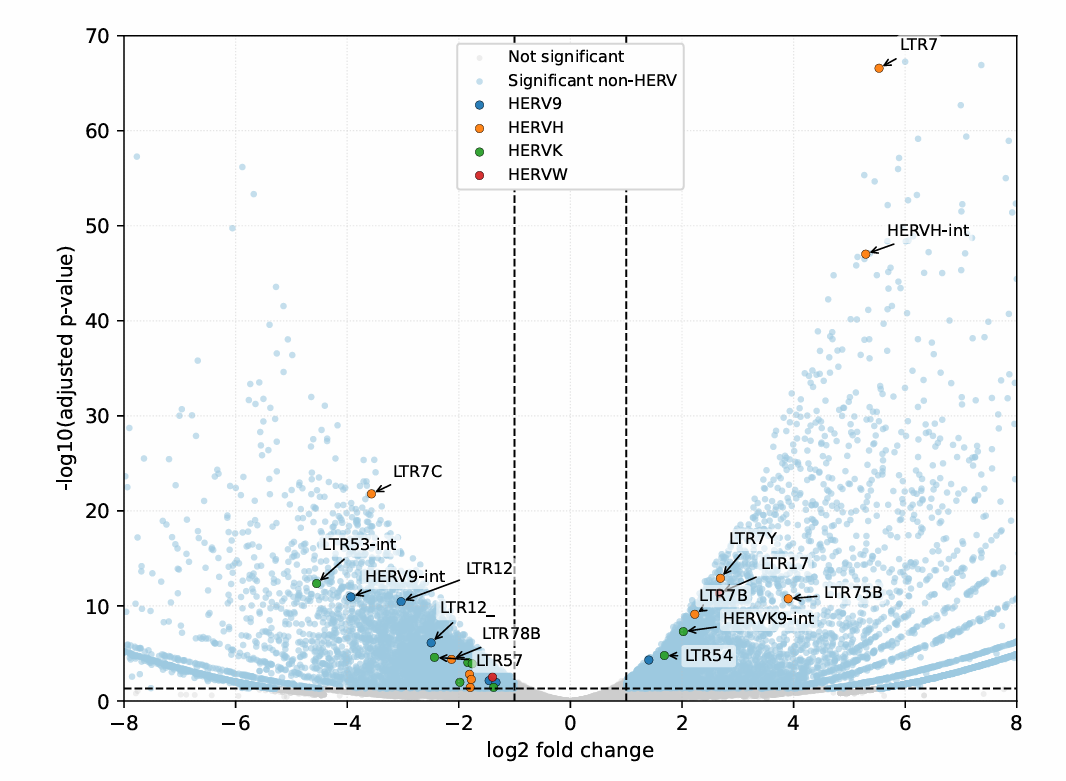
ERVmap
也是位点级,但使用的gtf是作者整理后的近全长(proviral)HERV位点(不覆盖大量碎片化/solo LTR等位点),相比telescope,比对更严格(舍弃多重比对读段,在高重复环境下尽量减少误计),而telescope将多重比对读段重分配(在HERV/HERVH这类高重复家族里很关键)
TE_Transcript_Assembly
主要分析对象是剪接位点(junction),把剪接事件和TE、基因外显子做整合统计(例如跨越各个剪接位点的读段数、每个外显子被TE覆盖的程度),最后把junction和外显子/TE交叠结果整合,统计每个junction对应的基因、类型(启动子/TSS一侧的边界、内部外显子边界、终止/TES一侧的边界)、两段关联TE的家族/类别、TE覆盖比例方向(与基因是同向还是反向)等。
前面的分析是说“哪个TE家族/位点有表达”,这个分析可以具体到“该TE是否参与了外显子结构”、哪个TE在什么基因的哪个位置发挥了结构作用(TE如何影响宿主基因结构/调控)