单细胞相关分析
other-other2025.11.18-2025.12.03研究进展
synapse网站数据
try 1:没用计算节点
直接读取会报错内存不够
import scanpy as sc
adata_b = sc.read_h5ad("synapseData/h5ad/a9/SEAAD_A9_RNAseq_DREAM.2025-07-15_2.h5ad")
print(adata_b)
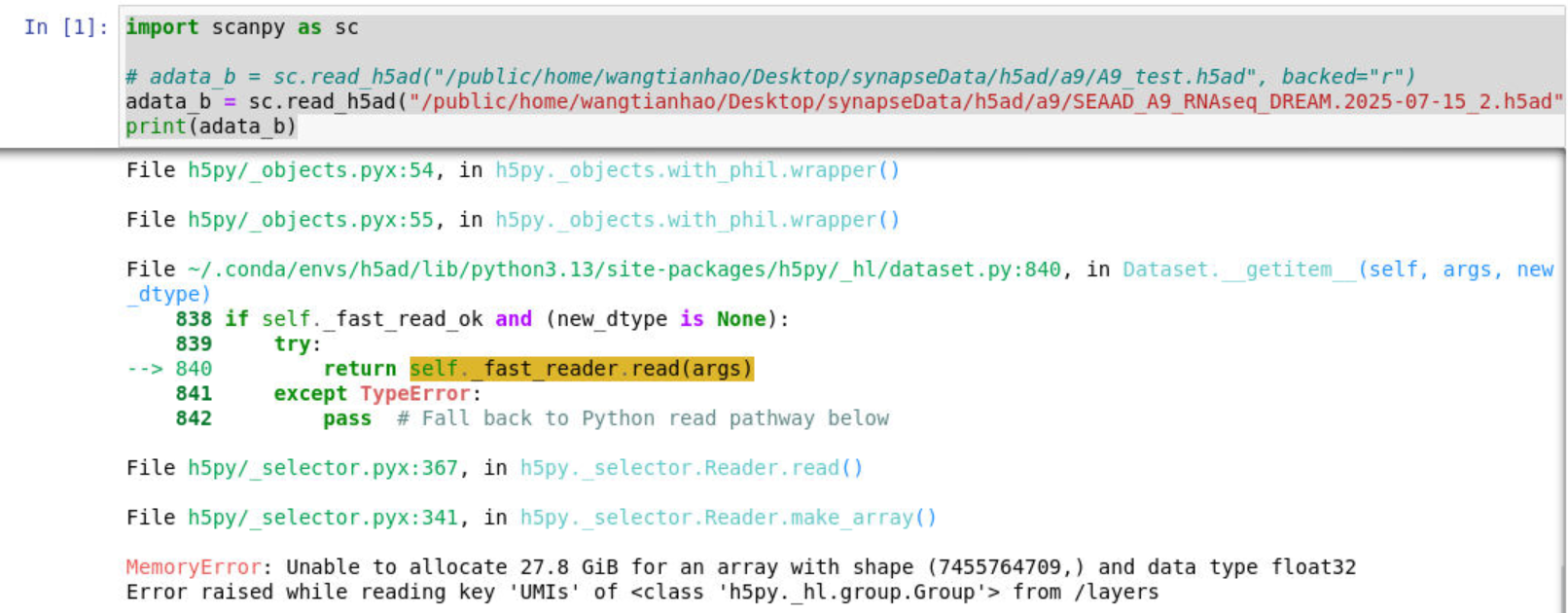
看了一下报错信息发现意思是在读/layers/UMIs这一层的时候,Python申请20多个G的内存失败,因此根据GPT的建议直接删掉/layers/UMIs这个数据,然后用read_h5ad(..., backed='r')读取,是不读进内存、只在磁盘上操作的方式(这个模式下很多操作不支持)
为了防止弄坏原文件,先cp一个副本A9_test.h5ad
import h5py
with h5py.File("synapseData/h5ad/a9/A9_test.h5ad", "r+") as f:
print("before:", list(f.keys()))
del f["layers"] # delete 'layers', avoid MemoryError
print("after:", list(f.keys()))
before: ['X', 'layers', 'obs', 'obsm', 'obsp', 'uns', 'var', 'varm', 'varp']
after: ['X', 'obs', 'obsm', 'obsp', 'uns', 'var', 'varm', 'varp']
import scanpy as sc
adata_b = sc.read_h5ad("synapseData/h5ad/a9/A9_test.h5ad", backed="r")
print(adata_b)

如图删去那层后读取成功,之后又因为Seurat分析时也需要大内存,同时因为这里我只是想验证一下这个h5ad能否正常进行读取和分析,因此按照GPT的方式,随机取一部分(这里是1000,如果更大的话可能运行时间很长)写成一个新的h5ad文件,并将其转换为Seurat可读的格式
# make a small h5ad file
import numpy as np
import anndata as ad
n_cells = adata_b.n_obs
n_keep = 1000 # randomly select some samples
idx = np.random.choice(n_cells, size=min(n_keep, n_cells), replace=False)
adata_small = adata_b[idx].to_memory()
adata_small.write_h5ad("synapseData/h5ad/a9/A9_test_small.h5ad")
# convert to Seurat type
from scipy.io import mmwrite
mmwrite("synapseData/h5ad/a9/counts.mtx", adata_small.X)
adata_small.obs.to_csv("synapseData/h5ad/a9/cell_metadata.csv")
adata_small.var.to_csv("synapseData/h5ad/a9/gene_metadata.csv")
上面我使用了python来转换格式,GPT还推荐R包SeuratDisk,但我发现官方提供的Rstudio好像不能自己安装新包,因此就没有用R
library(Matrix)
library(Seurat)
counts <- readMM("synapseData/h5ad/a9/counts.mtx")
cells <- read.csv("synapseData/h5ad/a9/cell_metadata.csv", row.names = 1)
genes <- read.csv("synapseData/h5ad/a9/gene_metadata.csv", row.names = 1)
counts <- t(counts)
rownames(counts) <- rownames(genes)
colnames(counts) <- rownames(cells)
obj <- CreateSeuratObject(counts = counts, meta.data = cells)
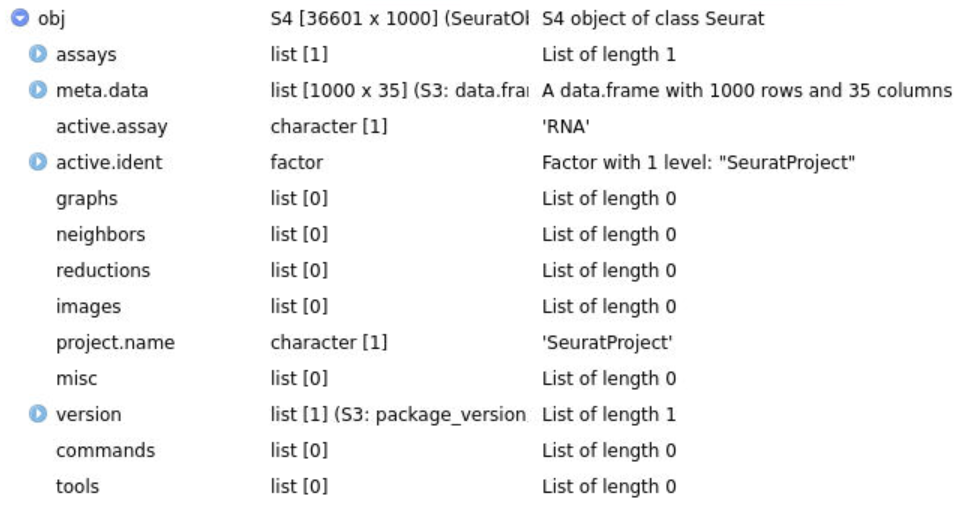
删掉的这层可能是原始计数矩阵(整数,没log、没归一化),因为是看用X那层生成的计数矩阵都是0~10的小数
try 2:使用计算节点
尝试使用STAR建索引
#!/bin/bash
#SBATCH -p DCU # 分区
#SBATCH -A chenjq # account
#SBATCH -J STAR_test # 作业名
#SBATCH -n 32 # 申请占用的CPU数,每个4G内存
#SBATCH --mem=128G # 显式说明申请多少内存
#SBATCH -o my_dcu_job.%j.out # 标准输出
#SBATCH -e my_dcu_job.%j.err # 标准错误输出
module load miniconda3/base
conda activate STAR_test
STAR \
--runThreadN 8 \
--runMode genomeGenerate \
--genomeDir /public/home/wangtianhao/Desktop/STAR_ref/test \
--genomeFastaFiles /public/home/wangtianhao/Desktop/STAR_ref/GRCh38.p14.genome.fa \
--sjdbGTFfile /public/home/wangtianhao/Desktop/STAR_ref/gencode.v49.annotation.gtf
使用的.fa/.gtf文件大小约3.1G(解压后),实测如果仅申请32G内存的话会溢出
读取h5ad文件:
#!/bin/bash
#SBATCH -p DCU # 分区
#SBATCH -A chenjq # account
#SBATCH -J h5ad_to_Seurat # 作业名
#SBATCH -n 16 # 申请占用的CPU数,每个4G内存
#SBATCH --mem=400G # 显式说明申请多少内存
#SBATCH -o my_dcu_job.%j.out # 标准输出
#SBATCH -e my_dcu_job.%j.err # 标准错误输出
module load miniconda3/base
conda activate h5ad
python -u /public/home/wangtianhao/Desktop/synapseData/h5ad_to_Seurat.py
h5ad文件大小约30G,如果申请256G内存会溢出,后来发现生产的mtx矩阵就有100多G
import os
import scanpy as sc
import scipy.sparse as sp
from scipy.io import mmwrite
def export_h5ad_to_seurat(h5ad_path, out_dir,):
os.makedirs(out_dir, exist_ok=True)
adata = sc.read_h5ad(h5ad_path)
X = adata.layers["UMIs"]
var = adata.var
if not sp.issparse(X):
X = sp.csc_matrix(X)
X_gc = X.T
mtx_path = os.path.join(out_dir, "counts.mtx")
mmwrite(mtx_path, X_gc)
features_path = os.path.join(out_dir, "features.tsv")
cells_path = os.path.join(out_dir, "cells.tsv")
var.index.to_series().to_csv(features_path, sep="\t", header=False, index=False)
adata.obs.index.to_series().to_csv(cells_path, sep="\t", header=False, index=False)
gene_meta_path = os.path.join(out_dir, "gene_metadata.csv")
cell_meta_path = os.path.join(out_dir, "cell_metadata.csv")
var.to_csv(gene_meta_path)
adata.obs.to_csv(cell_meta_path)
export_h5ad_to_seurat(r"/public/home/wangtianhao/Desktop/synapseData/h5ad/a9/a9_copy.h5ad", r"/public/home/wangtianhao/Desktop/synapseData/h5ad/a9/")
export_h5ad_to_seurat(r"/public/home/wangtianhao/Desktop/synapseData/h5ad/mtg/mtg_copy.h5ad", r"/public/home/wangtianhao/Desktop/synapseData/h5ad/mtg/")
因为觉得生成的文件都太大(约120-130w细胞、3.6w基因),不好用R进行后续分析,于是抽了3000个细胞做了一个小的h5ad(参考Seurat官方演示的数据集pbmc3k也是3000个细胞)
# 为了方便直接用了上面的slurm格式,感觉其实这里就不需要400G内存了
import os
import numpy as np
import scanpy as sc
import scipy.sparse as sp
from scipy.io import mmwrite
def make_seurat_test_dataset(h5ad_path, out_dir, n_cells=3000, random_state=0,):
os.makedirs(out_dir, exist_ok=True)
adata_b = sc.read_h5ad(h5ad_path, backed="r")
n_total = adata_b.n_obs
n_sample = min(n_cells, n_total)
rng = np.random.default_rng(random_state)
idx = rng.choice(n_total, size=n_sample, replace=False)
idx.sort()
adata_small = adata_b[idx].to_memory()
small_h5ad_path = os.path.join(out_dir, "test_subset.h5ad")
adata_small.write_h5ad(small_h5ad_path)
X = adata_small.layers["UMIs"]
var = adata_small.var
if not sp.issparse(X):
X = sp.csc_matrix(X)
X_gc = X.T
mtx_path = os.path.join(out_dir, "counts.mtx")
mmwrite(mtx_path, X_gc)
features_path = os.path.join(out_dir, "features.tsv")
cells_path = os.path.join(out_dir, "cells.tsv")
gene_meta_path = os.path.join(out_dir, "gene_metadata.csv")
cell_meta_path = os.path.join(out_dir, "cell_metadata.csv")
var.index.to_series().to_csv(features_path, sep="\t", header=False, index=False)
adata_small.obs.index.to_series().to_csv(cells_path, sep="\t", header=False, index=False)
var.to_csv(gene_meta_path)
adata_small.obs.to_csv(cell_meta_path)
make_seurat_test_dataset(
h5ad_path="/public/home/wangtianhao/Desktop/synapseData/h5ad/a9/a9_copy.h5ad",
out_dir="/public/home/wangtianhao/Desktop/synapseData/h5ad/a9_small/",
n_cells=3000,
)
make_seurat_test_dataset(
h5ad_path="/public/home/wangtianhao/Desktop/synapseData/h5ad/mtg/mtg_copy.h5ad",
out_dir="/public/home/wangtianhao/Desktop/synapseData/h5ad/mtg_small/",
n_cells=3000,
)
final try:h5ad to rds
因为上面生成的mtx有100多G,很吓人,所以看着能不能精简一下,因为在下面的GSE数据集中看到一个不过百M的rds格式文件读到R里后能有几个G大,就想着压缩成rds格式存储,在存储的时候使用了LoadH5Seurat函数,然后报错
经过亲测以及问gpt,发现R无法读取过大的h5ad文件,和什么格式无关,只是单纯因为数据太大而超过R数组索引的上限,必须要在python中分析。gpt给出的思路是:大规模QC、整合、降维、聚类等在python中做完,把感兴趣的几个cluster/子集导出成较小的数据集在R中分析(Seurat的可视化、美化图形、差异分析等)
最终从120w-130w细胞中抽取了2w个细胞作为较小的数据集
# conda activate h5ad
import os
import numpy as np
import scanpy as sc
import scipy.sparse as sp
from scipy.io import mmwrite
print("pkgs loaded")
def make_seurat_test_dataset(
h5ad_path,
out_dir,
n_cells=3000, # 抽取的细胞数
random_state=0,
):
os.makedirs(out_dir, exist_ok=True)
print(f"Reading big h5ad in backed mode: {h5ad_path}")
adata_b = sc.read_h5ad(h5ad_path, backed="r")
n_total = adata_b.n_obs
print(f"Total cells in dataset: {n_total}")
n_sample = min(n_cells, n_total)
print(f"Will sample {n_sample} cells as test dataset")
rng = np.random.default_rng(random_state)
idx = rng.choice(n_total, size=n_sample, replace=False)
idx.sort()
print("Subsetting and loading into memory ...")
adata_small = adata_b[idx].to_memory()
small_h5ad_path = os.path.join(out_dir, "test_subset.h5ad")
print("Writing small h5ad")
adata_small.write_h5ad(small_h5ad_path)
print(f"small h5ad: {small_h5ad_path}")
make_seurat_test_dataset(
h5ad_path="/public/home/wangtianhao/Desktop/synapseData/h5ad/a9/SEAAD_A9_RNAseq_DREAM.2025-07-15_2.h5ad",
out_dir="/public/home/wangtianhao/Desktop/synapseData/h5ad/a9_small/",
n_cells=20000,
)
make_seurat_test_dataset(
h5ad_path="/public/home/wangtianhao/Desktop/synapseData/h5ad/mtg/SEAAD_MTG_RNAseq_DREAM.2025-07-15.h5ad",
out_dir="/public/home/wangtianhao/Desktop/synapseData/h5ad/mtg_small/",
n_cells=20000,
)
因为我是先装的Seurat再装的SeuratDisk,发现第一次装的Seurat不能兼容任意版本的SeuratDisk,只能又开了一个环境装了SeuratDisk,再想装rhdf5时发现rhdf5不兼容这个版本的SeuratDisk,索性直接把rhdf5装在了之前的Seurat中
# conda activate r_seurat
library(rhdf5)
h5ad_file <- "/public/home/wangtianhao/Desktop/synapseData/h5ad/mtg_small/test_subset.h5ad"
obs_list <- h5read(h5ad_file, "/obs")
lens <- sapply(obs_list, length) # 每个元素的长度
n_cells <- max(lens) # 只取长度=细胞数的列
cell_cols <- names(lens)[lens == n_cells]
obs <- as.data.frame(obs_list[cell_cols], stringsAsFactors = FALSE)
rownames(obs) <- obs$exp_component_name
save(obs, file = paste0(h5ad_file, ".obs.RData"))
# conda activate r-seuratdisk
library(Seurat)
library(SeuratDisk)
h5ad_file <- "/public/home/wangtianhao/Desktop/synapseData/h5ad/mtg_small/test_subset.h5ad"
h5seurat_file = "/public/home/wangtianhao/Desktop/synapseData/h5ad/mtg_small/test_subset.h5seurat"
rds_file = "/public/home/wangtianhao/Desktop/synapseData/h5ad/mtg_small/mtg_small.rds"
load(paste0(h5ad_file, ".obs.RData"))
Convert(
h5ad_file,
dest = "h5seurat",
assay = "RNA",
overwrite = TRUE
)
obj <- LoadH5Seurat(
h5seurat_file,
meta.data = FALSE,
misc = FALSE
)
rownames(obs) <- obs$exp_component_name
obs <- obs[match(colnames(obj), rownames(obs)), , drop = FALSE]
obj <- AddMetaData(obj, metadata = obs)
saveRDS(
obj,
file = rds_file,
compress = "xz"
)
单细胞分析
基本原理
单细胞分析:在单个细胞的尺度上,一次性、高通量地测很多种分子信息,而且涉及到多种组学
- 高通量(high‑throughput):一次测几千、几万甚至几十万个细胞
- 分子信息(molecular profiling):细胞内有哪些种分子、每种有多少
- across modalities:RNA、染色质、蛋白、受体序列、空间位置
- 转录组(Transcriptomics):每个细胞里mRNA的丰度(每个基因大概多少转录本),比如Seurat分析scRNA‑seq数据
- 染色质开放程度(Chromatin accessibility):使用转座酶染色质可及性测序(ATAC-seq)、ChIP-seq等方法,看哪个DNA区域是开放的/被哪些蛋白占据
- 细胞表面蛋白(Surface protein expression):用带条形码的抗体测细胞表面标志,同时检测细胞表面的蛋白质信息与胞内转录组信息,相对转录组数据额外地加入了细胞表面蛋白的表达信息,可以更加深入地区分细胞异质性、更精确地挖掘特异性的细胞类型
- 适应性免疫受体组库(Adaptive immune receptor repertoire profiling):免疫细胞在识别哪些抗原、哪些克隆被扩增
- 空间信息(Spatial information):比如空间转录组,探究细胞在组织的哪个位置聚在一起、有什么相互影响
- 会有很多种分析方法,然后有人做了一些对比评测(benchmarking),告诉大家哪些方法在什么场景表现好、有哪些优缺点等等
- 尽量同时分析/整合多种模态(Multimodal),而不是只分析一种模态(Unimodal),比如RNA+ATAC,RNA+蛋白,RNA+空间位置…………
单细胞分析的三个任务:
-
聚类 & 细胞类型注释:将细胞按细胞类型聚类
- 聚类是一种无监督学习,每个细胞几千个基因的表达量,可以想成每个细胞是一个高维向量。具体来说
- 先做PCA,把几千个基因压缩到比如10–50个主成分
- 在主成分空间里,计算任意两个细胞之间的距离
- 对每个细胞,找到它最像的K个邻居(KNN图,k一般5–100)
- 通过一些划分算法来判断每个细胞属于哪个cluster
-
标志基因(marker):在某一类细胞里明显高表达,而在其他细胞里表达低或没有的基因。具体来说
- 先看某个cluster,与其他所有cluster比较
- 进行差异表达分析(t-test/Wilcoxon rank-sum test)
- 选出在这个cluster表达比例高,在其他表达比例低,且logFC明显为正、p值显著的基因
-
细胞注释的三个层次/步骤
-
自动注释
- Classifier-based:对于每个聚类,根据标志基因表达模式,训练一个分类器,例如Garnett/CellAssign/CellTypist/Clustifyr等
- 映射到参考图谱(Reference mapping):把数据投到一个已经注释好的大图谱上,例如scArches/Symphony/Azimuth等
- 手动注释:直接查看每个聚类的标志基因,再查阅相关文献,或者参考经典marker
- 验证注释的可靠性:例如marker是否符合生物学常识、同一细胞类型是否只出现在一个cluster、不同样本之间能不能对应起来等,可能还会用细胞谱系、轨迹等信息辅助判断
-
自动注释
- 聚类是一种无监督学习,每个细胞几千个基因的表达量,可以想成每个细胞是一个高维向量。具体来说
-
连续过程 & 轨迹 + RNA velocity + 谱系追踪
-
轨迹推断(trajectory inference):很多生物过程不是“划分成几类”就够了,比如细胞的分化过程、T细胞激活-记忆T细胞状态改变的过程,是一个连续的轨迹,而不是简单的几个聚类。轨迹推断就是在表达空间里找到一条(或几条)“路线”,把细胞按照生物过程的顺序排成一条线(伪时间pseudo time)
- Cluster approach(聚类 → 线连接):先聚类,再看cluster之间的拓扑连接关系,用最小生成树MST连接;每个cluster是一个节点,MST把它们连起来,得到一条或分叉的路径
- Graph approach(图论方式):先在细胞间构图,再用图的方法(比如PAGA)推断cluster之间的连通关系和方向,更适合复杂拓扑(多分支)
- Manifold-learning based(流形学习):在降维空间里找一条“光滑曲线”,把cluster串起来,或者说,在点云之间画一条最顺最合理的“生长路径”,如Slingshot
- Probabilistic frameworks(概率框架):基于扩散过程,给每个细胞一个“到起点的距离”的概率度量 → 伪时间,如DPT
需要注意的是,推出来的轨迹不一定有生物学意义,需要结合实验背景判断;对稳态系统/多种复杂动力学,传统伪时间有时不适用
-
RNA velocity:用“未剪接RNA(u)”和“已剪接RNA(s)”的比例,来推断:这个基因在这个细胞里,是在“被进一步打开/即将表达更多”,还是在“关上/即将表达更少”
- 根据测序时读段在外显子/内含子上的位置,统计每个细胞、每个基因的unspliced和spliced reads数量
- 假设在某个时间点,基因表达处于稳定状态,可以估计降解速率γ:
ds/dt=βu-γs,当ds/dt=0时用u和s估计γ - 用unspliced数量+估计的γ,推断下一时刻的spliced数量,如果u很高,就说明正准备大量剪接成s,s会升高,基因转录更活跃;如果u低但s高,说明转录已停止,只剩下降解,s会降低,基因转录更沉默
- 对每个细胞,找到“最像它下一步状态”的其他细胞:在UMAP/tSNE图上,就画出一根箭头指向“未来的位置”(velocity vector)
本质上就是在原来的表达空间上,叠加一个“流场”,说明细胞群整体在往哪里演化
-
谱系追踪:从基因组突变/条形码角度推断“谁是谁的后代”
- 前瞻追踪(Prospective lineage tracing):在起始细胞引入一个可遗传的标记(例如条形码/CRISPR 可编辑位点),后代细胞都会带着这个标记;标记不断累积变化,最后后期测序这个标记来还原“家谱树”,例如软件Cassiopeia
- 回顾追踪(Retrospective lineage tracing):不事先刻条形码,直接利用自然发生的体细胞突变,来推断细胞之间的亲缘关系,类似构建“细胞的系统发育树”
DNA条形码(DNA barcoding):一条某个物种特有的、且在种内稳定遗传的DNA序列,标识DNA序列和生物物种之间对应的关系,可用于物种快速鉴定
-
轨迹推断(trajectory inference):很多生物过程不是“划分成几类”就够了,比如细胞的分化过程、T细胞激活-记忆T细胞状态改变的过程,是一个连续的轨迹,而不是简单的几个聚类。轨迹推断就是在表达空间里找到一条(或几条)“路线”,把细胞按照生物过程的顺序排成一条线(伪时间pseudo time)
-
揭示机制:在完成分类和注释之后,进一步探究为什么会有区别,是什么因素导致了差异
-
差异表达:单细胞计数数据常用负二项分布建模,差异比较有两种思路:
- Sample-level(pseudobulk):把同一类型的所有细胞在同一样本里加和,得到一个样本级的“pseudo-bulk count”,然后像bulk RNA-seq(对一群细胞的测序分析)那样,用edgeR/DESeq2/limma做差异分析,适合做病例-对照(处理“每个病人/样本”为单位的变异,统计更严谨)
- 单细胞级(Cell-level):直接把每个细胞当一个观测,用广义线性混合模型建模,能更细致建模“零膨胀”等单细胞特性,但计算量大,例如工具MAST
-
基因集富集:哪些通路/TF活性在变化。有一些工具
- PROGENy:不用“通路上的所有基因”,而用“下游footprint基因”推断通路活性,更适合scRNA
- DoRothEA:给TF一组target基因,算每个TF的活性(而不是只看TF自己表达)
- Pagoda2:专门为scRNA设计的富集分析框架
-
细胞比例变化:不同条件之间,某一种细胞类型的比例有没有变化
- 比较简单的情况:统计各样本中某细胞类型的数量,做Poisson回归或非参数检验
- 更复杂时:多个细胞类型沟槽一个组成(composition),和样本总细胞数相关,需要考虑组成数据(compositional data)的特性——总和固定,增加一个类型的比例必然导致其他类型比例下降,例如scCODA/tascCODA等方法用贝叶斯模型来处理这个问题
- DA-seq/MILO等方法更进一步,不是只对“人工划的cluster”做丰度比较,而是对局部邻域/连续空间做DA分析
DE(Differential expression):两组样本的同一细胞类型的基因表达差异分析
DA(Differential abundance):两组样本的同一细胞类型的丰度差异分析
-
扰动建模(perturbation):用CRISPR-Cas9在细胞中敲除或敲低很多基因(每个细胞带一个gRNA条形码);同时做单细胞RNA测序,从而知道这个细胞的gRNA是哪个,以及全转录组表达情况;最后分析某个基因敲除后对整个转录组的影响,进而对对细胞类型的影响。有一些工具
- Augur:根据“条件(刺激-对照)能多好地被分类器区分”,找到对扰动最敏感的细胞类型
- scGen:用生成模型预测“如果对一个细胞做某种扰动,转录组会如何变化”
- Mixscape:在CRISPR中,区分真正被编辑的细胞和没编辑上的细胞
-
细胞–细胞通讯:一个细胞分泌配体(ligand),另一个细胞表面有受体(receptor),进而激活下游通路
从表达数据里推断配体和受体的相互作用:先有一个ligand–receptor配对数据库,对每一对细胞类型A(发送者)和细胞类型B(接受者),看A是否表达配体、B是否表达受体,再看B是否出现了该通路的下游目标基因激活
- LIANA:一个框架,整合多个通讯工具的输出
- NicheNet:把ligand–receptor与下游基因调控网络结合起来,预测哪个配体可能解释某些目标基因的上调
-
差异表达:单细胞计数数据常用负二项分布建模,差异比较有两种思路:
单细胞转录组(Transcriptomics):
-
实验流程:样本采集 → 细胞解离 → 单细胞分离 → RNA提取 → 建库测序 → 初级数据分析 → 高级数据分析
-
两种主要建库方式:
- 基于微孔板(Plate-based):每个细胞手工分到96/384孔板,每个孔独立扩增测序;深度高、可以测更多基因,但通量较低
-
液滴法(Droplet-based):以10x为代表,细胞和带有条形码的beads在油相中形成一个个液滴。每个bead上标识cell barcode(这个bead/液滴/细胞的id)和UMI(这个转录本分子的id)
cell barcode用来区分“这个reads来自哪个细胞”,UMI用来区分“这个reads来自哪个原始转录本分子”
- 完成测序后,cellranger负责读入FASTQ文件(原始读段)并比对到参考基因组,之后按基因+细胞条形码+UMI计数,并做cell calling(区分真实细胞和背景液滴)最后输出结果
-
matrix.mtx:一个稀疏矩阵,行是基因,列是细胞,值是UMI计数 -
genes.tsv/features.tsv:每行对应一个基因/feature,第一列是基因id(如ENSG0000…),第二列是基因symbol(如ACTB),第三列是feature type(如Gene Expression/Antibody Capture/Peak等) -
barcodes.tsv:每行一个细胞条形码,顺序与矩阵列一一对应,例如AAACCTGAGAAACCAT-1这样的字符串
注:Seurat里用
Read10X(...)就是自动读这三样东西,然后CreateSeuratObject就会把它们组合成一个对象 -
-
两种主要建库方式:
-
质量控制:理想情况下,每个液滴里只有一个完好细胞
-
实际操作中存在的问题
-
低质量细胞(死亡/破膜):细胞破裂,细胞核内mRNA流失,只剩少量线粒体RNA,导致检测到的基因数很少、UMI总数低、线粒体基因(以
MT-开头的)比例高 - 背景RNA:样本制备时,部分细胞破裂,RNA游离进溶液里,被其它液滴吸入,导致一些标志基因在不该出现的细胞里也有一点表达。有一些工具比如SoupX、CellBender,可以估计背景成分,校正计数矩阵
- 双细胞(Doublets):一个液滴里装了两个细胞,导致测得的表达是两个细胞的叠加。scDblFinder通过“模拟双细胞表达,和真实细胞比较”来识别
-
低质量细胞(死亡/破膜):细胞破裂,细胞核内mRNA流失,只剩少量线粒体RNA,导致检测到的基因数很少、UMI总数低、线粒体基因(以
- 中位数绝对偏差(median absolute deviation, MAD):偏离中位数5个MAD的细胞被定为异常细胞(极端值outlier)。标准差对极端值很敏感,MAD更稳健
-
实际操作中存在的问题
-
归一化(normalization):想要不同细胞之间可比较
- 简单log归一化(Shifted log transformation):先按每个细胞的测序深度“拉平”总量,再取对数压缩极端值
- scran normalization/delta method:用池化的方式估计size factor,比简单“每个细胞总counts”更稳,特别适合批次校正任务
- 基于NB模型的残差变换(Analytic Pearson residuals):计算每个基因、每个细胞的Pearson残差,残差大则表示该细胞这个基因的表达显著高于“技术噪声”预期,这些残差作为新矩阵,方差更稳定,适合找高变基因和稀有基因型
注:在Seurat里,
NormalizeData是简单log,SCTransform类似Analytic Pearson residuals -
数据校正:
-
技术偏差:
- 批次效应(batch effect):不同实验批次/不同平台的系统差异
- 技术上的抽样波动(count sampling effects):同样表达水平,测序深度不同
- 生物混杂因素(biological confounding):细胞周期阶段不同会牵动一大批基因,进而影响聚类
-
常见方法:
- 线性嵌入模型(Linear-embedding models):例如Harmony,在PCA等低维空间里做对齐,适合“结构比较简单”的整合任务
- 深度学习方法:例如基于变分自编码器的scVI、进一步改进的scANVI,适合复杂的大规模整合任务
- 细胞周期校正(Cell cycle correction):比如Seurat内置的方法、Tricycle
-
技术偏差:
-
特征选择与降维:不可能拿2万个基因直接聚类,先选出信息量大的500–5000个基因(高变基因),再做 PCA/tSNE/UMAP
-
特征选择的指标:
- 平均表达量(mean):非常低的基因大多是噪声,不考虑
-
变异度/离散度(dispersion/coefficient of variation, CV):
CV=标准差/均值,Dispersion对CV做一些标准化处理,减弱均值的影响 - 偏差(Deviance):在负二项分布模型下,某基因在不同细胞之间的表达是否偏离“只有技术噪声”的预期,偏差越大,越可能反映真实生物差异
-
降维方法:
- PCA:线性方法,捕捉全局变化,几乎是所有pipeline的第一步
- t-SNE:非常擅长展示局部结构(细小聚类),但对参数敏感
- UMAP:在稳定性、保持局部和全局结构等方面表现好
-
特征选择的指标:
单细胞表观组(epigenomics):直接探究“哪个增强子/启动子在哪种细胞类型是打开的”,这是转录调控的核心
-
基本原理:
- ChIP-seq/CUT&Tag/CUT&RUN:用特异抗体拉下带某种修饰或TF的DNA片段,测序,就知道TF/修饰在哪里富集
-
ATAC-seq:用Tn5转座酶插入接头,只在开放染色质(没有紧密绕在核小体上)插入,后面测序这些片段,重新比对到基因组上,就知道哪些位置开放了,插入的越多说明越开放/越有富集
- 切点(cut/cut site):Tn5在某个碱基附近插入的事件
- peak:把基因组位置作X轴、每个位置的cut数量作Y轴,画一个曲线,曲线的峰点就是peak。一个peak对应一个短的基因组区间,比如200–1000bp,在这个区间里cuts明显高于周围背景且有统计学意义
很多peaks对应启动子、增强子、绝缘子(CTCF位点)等各种功能调控元件
在实际应用中,常统计每个细胞中在每个peak里有多少cuts,得到一个cell×peaks的计数矩阵,对这个矩阵作归一化/PCA等分析
- 单细胞版的scChIP/scATAC:把这些流程和微流控/索引技术结合,对每个细胞单独建库。scChIP比较难做,深度低;scATAC相对成熟,单细胞分析多用scATAC
-
常见修饰:
- H3K4me1/H3K27ac:增强子
- H3K4me3:启动子
- H3K36me3:转录中的基因区域
- H3K27me3:Polycomb抑制相关标记(被Polycomb复合体PRC1/PRC2调控的区域),往往是发育调控基因、需要被沉默或待命的基因
- H3K9me3:异染色质(heterochromatin)
-
scATAC/scChIP分析的优势
- 能区分调控原因(比如哪个增强子更活跃),而不仅是结果(哪个基因高表达)
- 能识别新的顺式调控元件(CRE):很多增强子远离基因,RNA看不到
- 和scRNA联合,可以构建TF–CRE–Gene的调控网络
-
scChIP-seq和scATAC-seq的流程:读入数据 → 质量控制 → 定义特征features → 降维聚类 → 细胞注释 → 可视化 → 深入解读(motif、footprinting、整合RNA等)
-
scATAC的QC指标
- Total fragment counts:类似scRNA的总counts,太低就是低质量
- TSS enrichment:转录起始位点附近读段的富集程度,开放染色质在TSS周围通常有明显峰,比值高则信噪比好
- Number of features per cell:类似检测到的peak数,相当于scRNA的基因数
- Nucleosome signal:单核小体长片段与无核小体短片段的比值,比值过高或过低都可能是异常
- Reads in peaks fraction:落在peak区域的读段在全基因组中的占比,偏低则噪声大
- Blacklist fraction:落在ENCODE定义的“黑名单区域”的reads比例(这些区域容易产生假阳性信号)
- Doublet检测:可用scDblFinder、AMULET等,通过检测“某个基因组位置>2个reads”的情况判断doublet
-
scATAC的QC指标
单细胞表观组的三个任务:
-
scATAC的聚类和注释:不能直接拿peak-count矩阵做PCA(太稀疏)
- 常用:
- Latent Semantic Indexing, LSI:类似于文本挖掘,peak相当于词,细胞相当于文档,先做TF-IDF(一种归一化方法),再做SVD,例如Signac、ArchR
- Latent Dirichlet Allocation, LDA:把peak看成“topic”,每个细胞是多个topic的混合,例如cisTopic
- Spectral embedding:使用谱嵌入做降维,例如snapATAC
- Normalization + Binarization:常见做法是把peak counts转成二值(开放/不开放),忽略read深度差异,减少技术噪声,不过会丢掉“开放程度强弱”的信息
-
差异可及区域(DARs)和注释:像差异表达一样,对比两个细胞型,找到差异开放的peaks作为DARs,用DESeq2/edgeR等模型,需要考虑混杂因素(confounder),例如测序深度/批次/线粒体读段比例/细胞类型阶段等
DARs对应的CRE:
- 用GREAT/LOLA/GIGGLE等工具做富集分析(看这些CRE富集在哪些TF motif/哪些基因附近)
- 和目标基因关联(最近基因/loop等),算gene activity score,做细胞类型注释
- 常用:
-
TF motif & footprinting
- chromVAR:把“哪些peak含有某个motif”信息加总起来,给每个细胞一个这个TF motif的活性分数
-
粗略流程:
- 对每个motif,找出所有含有它的peaks
- 对每个细胞,算这些peaks的总reads/counts
- 根据GC含量和总体可及性,为每个motif找一组背景peaks,算背景期望
- 比较实际和背景,得到bias-corrected deviation(Z-score)
- 形成motif×cell矩阵,用于细胞聚类、motif差异可及性分析和推断TF活性/协同作用
- Footprinting:看motif位置周围的cutpattern:真正有TF结合的地方,转座酶/酶切会被挡住,让motif位置出现“凹槽”(cut掉两侧,中间低)。可以更有力地证明“这个TF在这个细胞型中真的在DNA上起作用,而不仅仅是motif存在
-
整合scATAC和scRNA:取决于是同一个细胞测了多模态,还是不同细胞测的不同模态
- Paired integration:同一细胞有RNA+ATAC,如10xmultiome/sci-CAR等。使用MOFA+(多模态因子分析),同时建模多模态
- Unpaired integration:一批细胞测RNA,另一批测ATAC。使用SCOT/UnionCom/GLUE等,目标是对齐两种模态的manifold,尽量减少信息损失
- Mosaic integration:有的细胞测一部分模态,有的测另一部分,有的测全部如totalVI/MultiVI(对CITE-seq/multiome)建联合模型。使用Stabmap/Multigrate等可以把单模态和多模态数据一起映射到一个共享空间,并补全缺失模态
以下以R包Seurat为例
pbmc3k数据的分析
library(Seurat)
library(SeuratData)
library(tidyverse)
InstallData("pbmc3k")
# 或直接下载http://seurat.nygenome.org/src/contrib/pbmc3k.SeuratData_3.1.4.tar.gz,之后本地安装install.packages("C:\\Users\\17185\\Downloads\\pbmc3k.SeuratData_3.1.4.tar.gz", repos = NULL, type = "source")
InstallData("ifnb")
# 或直接下载https://seurat.nygenome.org/src/contrib/ifnb.SeuratData_3.1.0.tar.gz,之后本地安装install.packages("C:\\Users\\17185\\Downloads\\Compressed\\ifnb.SeuratData_3.1.0.tar.gz", repos = NULL, type = "source")
载入数据
library(pbmc3k.SeuratData)
data("pbmc3k")
pbmc <- UpdateSeuratObject(object = pbmc3k)
认识Seurat对象:
pbmc # 有多少个细胞和features,以及当前的数据类型(RNA)
slotNames(pbmc) # 有哪些slot
head(pbmc@meta.data) # nCount_RNA和nFeature_RNA是后面质控要用的两个基础指标
常用的slot:
-
@assays:表达矩阵、归一化结果等 -
@meta.data:细胞的各种信息(行为细胞) -
@active.ident:当前的“分组标签”(后来聚类就写在这) -
@reductions:PCA/UMAP等降维结果
质控(QC)
加上线粒体比例:PercentageFeatureSet
- 标记“线粒体基因比例”,用来排除可能是坏掉/应激严重的细胞
pbmc[["percent.mt"]] <- PercentageFeatureSet(
pbmc,
pattern = "^MT-" # 人类线粒体基因以 MT- 开头
) # 在 meta.data 里新建一列叫 percent.mt
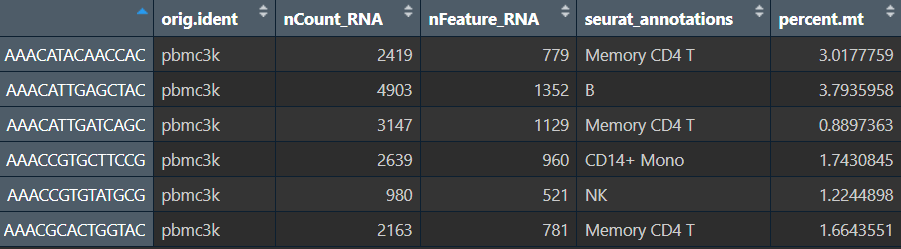
head(pbmc@meta.data)
现在meta.data就有三列关键QC指标了:
-
nFeature_RNA:这个细胞检测到多少基因 -
nCount_RNA:这个细胞一共多少UMI -
percent.mt:线粒体reads占百分之多少
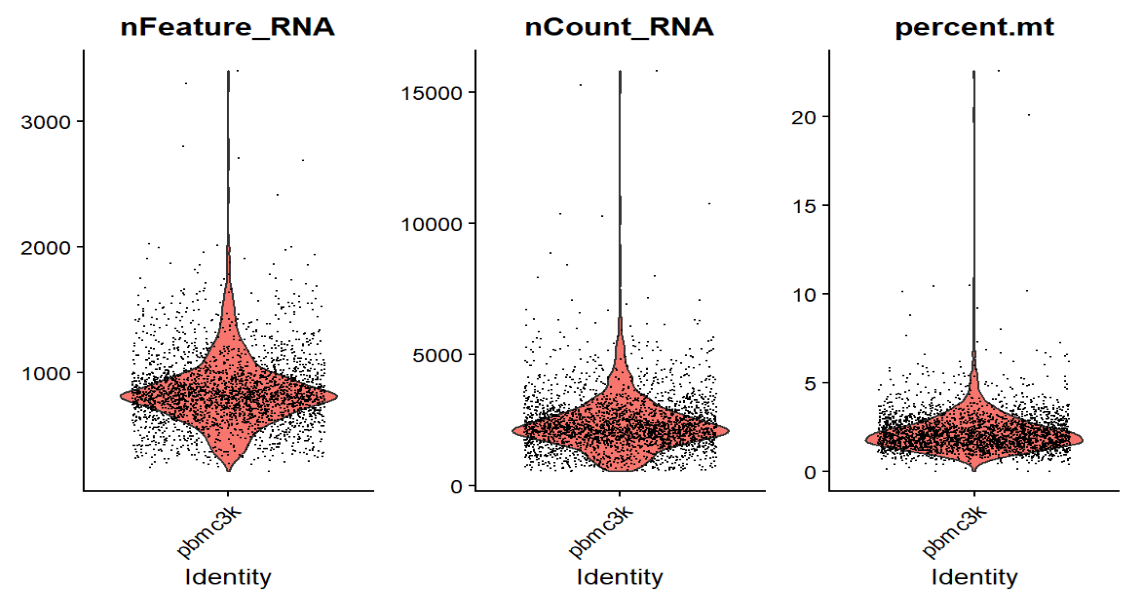
看QC图:VlnPlot+FeatureScatter
- 看看这些指标的分布,大致选过滤阈值
# 小提琴图
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
# 两个散点图
FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
VlnPlot:画小提琴图,每个点是一细胞
-
features:要画的列名(必须在meta.data或Assay中存在) -
group.by:按什么分组画(还没聚类就默认一组)
FeatureScatter:x/y都是meta.data里的列名
- 第一张:总UMI - 线粒体比例
- 第二张:总UMI - 检测基因数
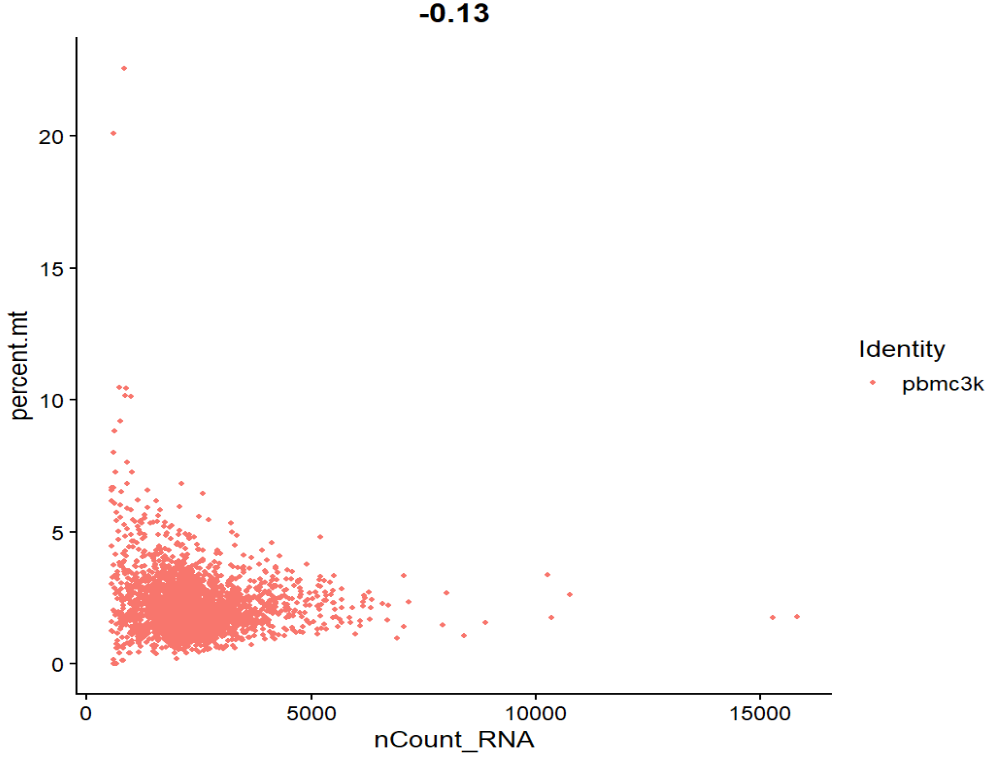
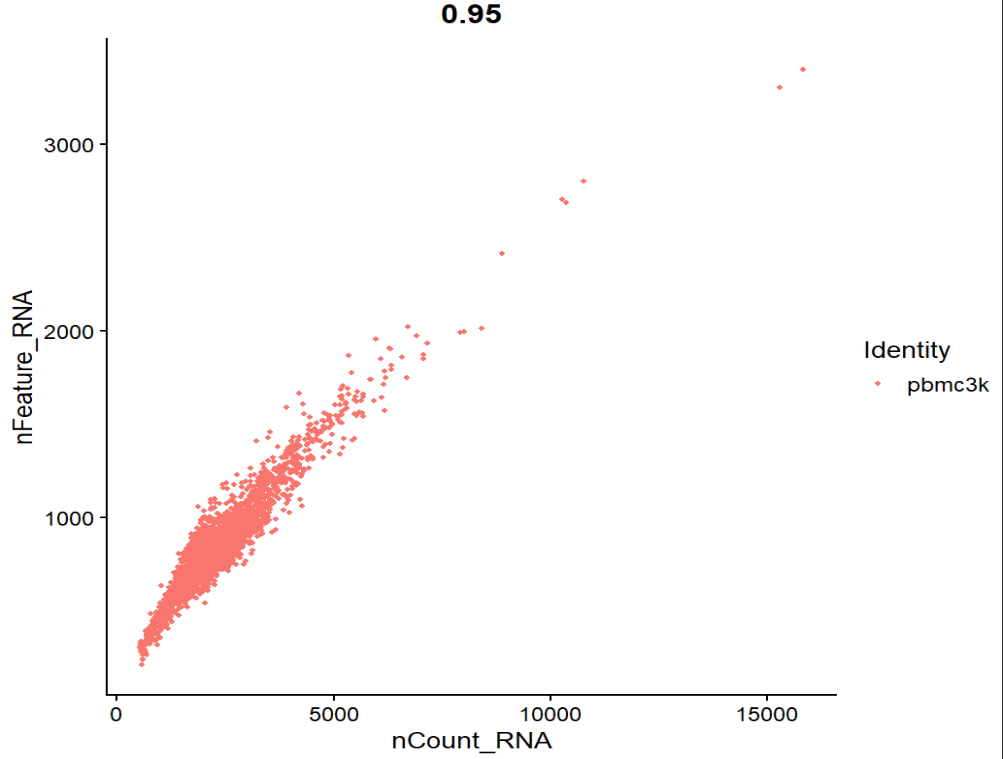
- 右下角那种nCount很高但nFeature不升反降的点,往往是doublet/奇怪细胞
- percent.mt特别高的一群,很可能是坏细胞
按阈值过滤:subset
- 把“很可疑”的细胞直接丢掉,减少后面噪音
pbmc <- subset(
pbmc,
subset = nFeature_RNA > 200 & # 阈值根据上面画的图确定
nFeature_RNA < 2500 &
percent.mt < 5
)
归一化/高变基因/标准化
有两条路线:
- 经典路线:
NormalizeData→FindVariableFeatures→ScaleData - 推荐的新路线:一次性用
SCTransform
NormalizeData:把不同细胞的测序深度拉到同一量级
- 不同细胞测到的reads/UMI总数不一样,直接比较counts不公平
具体做法:
- 每个细胞按总UMI做缩放(库大小归一化)
- 对缩放后的数值取log1p(稳定方差)
pbmc1 <- NormalizeData(
pbmc,
normalization.method = "LogNormalize", # log(x/UMI*10^4 + 1)
scale.factor = 1e4, # 把每个细胞的总表达量缩放到10^4左右
verbose = FALSE
) # pbmc1[["RNA"]]@data里存的就是log归一化后的表达矩阵
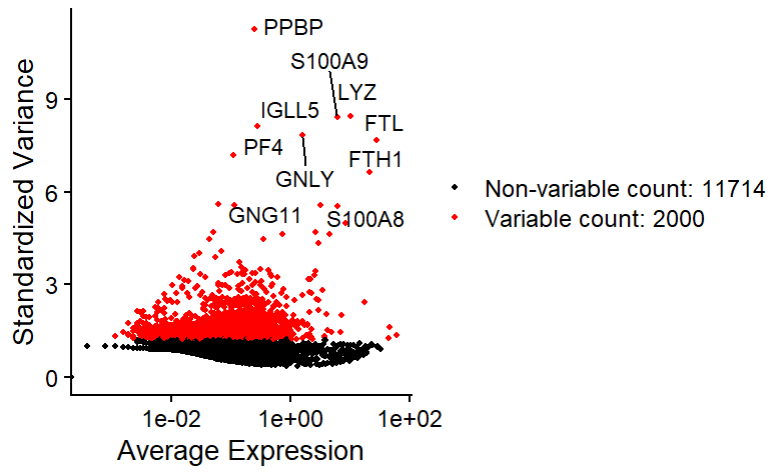
FindVariableFeatures:找信息量最大的那几千个基因
- 几万个基因里,真正对区分细胞类型有帮助的只有几千个
- 先把这些“高变基因(HVG)”选出来,后面PCA/聚类只用它们,减少噪声
pbmc1 <- FindVariableFeatures(
pbmc1,
selection.method = "vst", # 官方推荐的默认方法(variance-stabilizing transformation)
nfeatures = 2000 # 选多少个HVGs(通常2000–3000)
)
# 查看选出来的基因
top10 <- head(VariableFeatures(pbmc1), 10) # 返回HVGs的基因名向量(前10个)
VariableFeaturePlot(pbmc1) # 横轴=均值,纵轴=标准化后的离散度,红色点是HVG
LabelPoints(VariableFeaturePlot(pbmc1), points = top10, repel = TRUE) # 给指定的点加标签
ScaleData:做Z-score标准化和协变量回归(可选)
- 每个基因做标准化:让不同基因有可比性
- (可选)对某些技术/生物因素做回归,比如线粒体比例、细胞周期
pbmc1 <- ScaleData(
pbmc1,
features = VariableFeatures(pbmc1), # 要scale的基因集合,通常只对HVG做就够了
vars.to.regress = c("percent.mt", "nCount_RNA"), # 做一个简单线性回归,把这些变量对表达的线性影响减掉
verbose = FALSE
) # pbmc1[["RNA"]]@scale.data存的就是这个Z-score矩阵(行为基因,列为细胞)
SCTransform:用一个负二项GLM模型,同时完成归一化+方差稳定+回归协变量,比上面三步更严谨。官方建议优先使用 SCTransform,尤其是样本多、噪音复杂时
pbmc2 <- SCTransform(
pbmc,
vars.to.regress = "percent.mt",
verbose = FALSE
)
注:vars.to.regress参数的设置
- 本质上是把指定的变量(比如
nCount_RNA、percent.mt)对该基因表达的线性影响减掉,这些变量是我们不想要的协变量,希望它没有线性影响 -
nCount_RNA是每个细胞的总UMI数,反映:技术因素(测序深度、捕获效率),也可能反映一些生物因素(细胞大小、活跃程度),在第一种方法中,因为已经做了NormalizeData,理论上已经按总counts做过一次“库大小归一化”了。如果在实践中,残余的深度效应仍然明显(比如UMAP上按nCount_RNA上色会出现梯度),就可以在ScaleData里回归nCount_RNASCTransform内部已经用负二项GLM建模了测序深度等因素,一般不需要(也不建议)再额外回归nCount_RNA/nFeature_RNA。可以在SCTransform里只回归一些你确实不希望作为主信号的生物/技术变量,比如percent.mt或细胞周期得分 -
percent.mt是线粒体reads占比。即使已经在QC阶段过滤掉特别极端的高mt细胞,剩下细胞里仍可能存在mt稍微偏高的那拨,在PCA里变成前几个PC。因此为了避免让线粒体比例主导聚类结果,通常会指定这个
PCA
RunPCA:把几千个HVG的维度压缩成几十个主成分
pbmc3 <- RunPCA(
pbmc2,
features = VariableFeatures(pbmc2), # 用于PCA的基因,一般就是HVG
npcs = 50, # 要算多少个PC(常用50)
verbose = FALSE
)
PCA结果存在pbmc@reductions$pca里,包含:
-
@cell.embeddings:每个细胞在PC1、PC2…上的坐标 -
@feature.loadings:每个基因在各PC上的载荷(贡献度)
看PCA结果:VizDimLoadings、DimPlot、DimHeatmap、ElbowPlot
VizDimLoadings(pbmc3, dims = 1:2, reduction = "pca")
DimPlot(pbmc3, reduction = "pca")
DimHeatmap(pbmc3, dims = 1:10, cells = 500, balanced = TRUE)
ElbowPlot(pbmc3, ndims = 50)
-
VizDimLoadings:看PC上贡献最大的基因(帮助理解每个PC在分什么)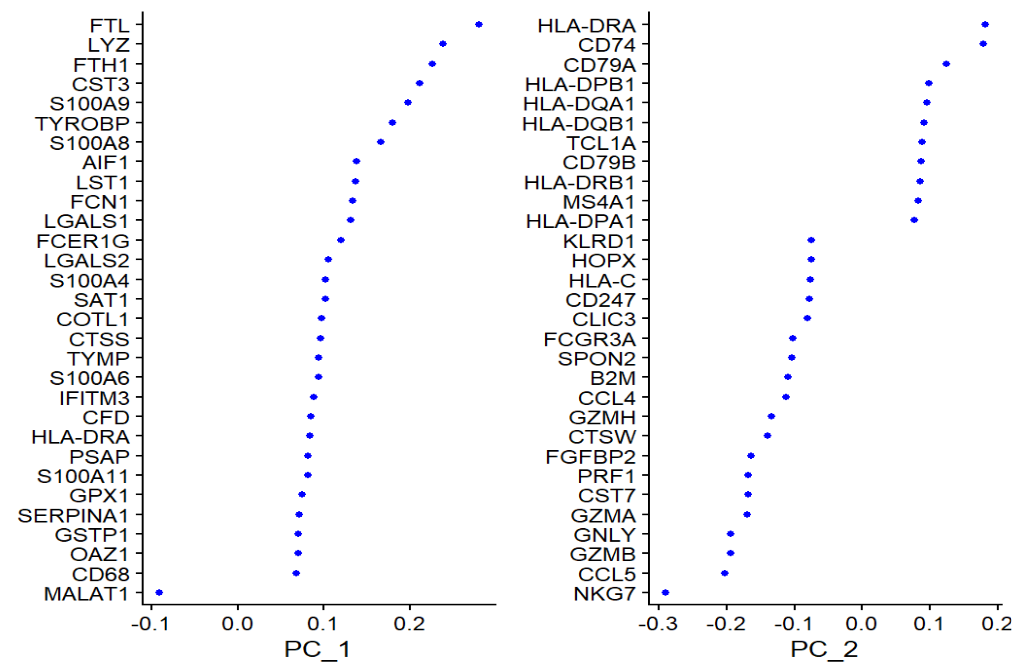
-
DimPlot(..., reduction = "pca"):画PC1-PC2的散点图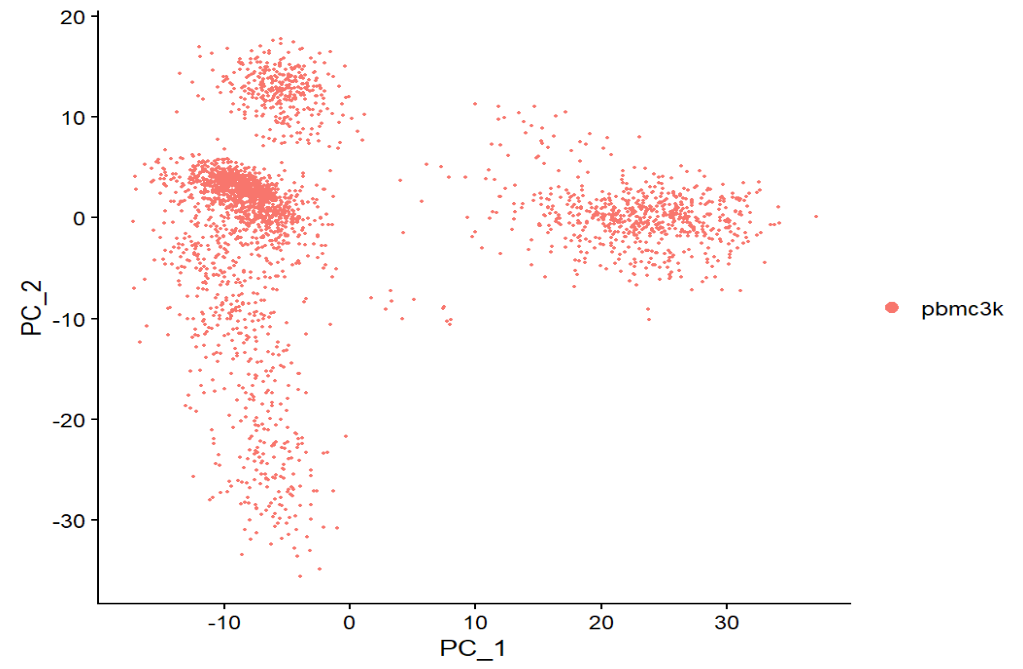
-
DimHeatmap:看某几个PC对哪些细胞/基因区分度大
-
ElbowPlot:选多少个PC用于后面的聚类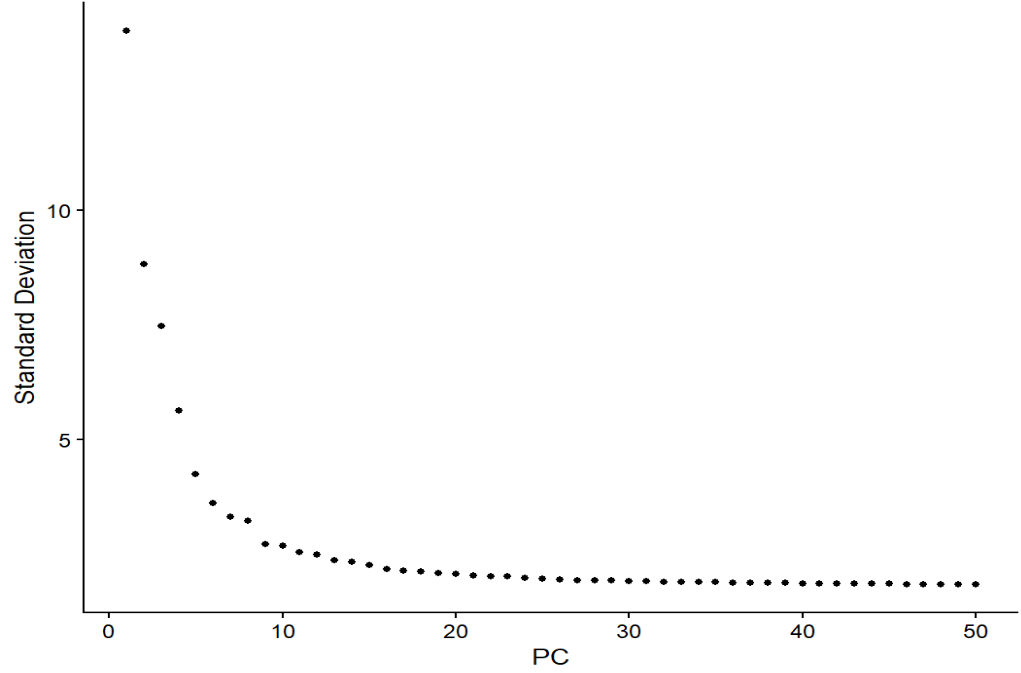
- 横轴PC序号,纵轴每个PC的变异解释度
- 找“肘部(elbow)”的位置(即曲线的弯曲部位),一般10-20左右,看具体图而定
构图 & 聚类
FindNeighbors:构建KNN图
- 在PCA空间里找每个细胞的K个近邻,形成一个图,后面的聚类就在这个图上做
pbmc4 <- FindNeighbors(
pbmc3,
dims = 1:10, # 使用哪些PC(根据上面ElbowPlot)
k.param = 20 # 每个点找多少邻居,默认20,过小会太稀疏,过大会挤在一起
)
图结构存到pbmc@graphs,一般至少有两个:
-
RNA_snn:shared nearest neighbor graph -
RNA_nn:最近邻图
FindClusters:在图上进行Louvain/Leiden,用图算法把细胞分成几个cluster
pbmc4 <- FindClusters(
pbmc4,
resolution = 0.5,
algorithm = 1 # 默认1 = Louvain,4 = Leiden
)
-
resolution:控制cluster数量的关键参数,越大则cluster越多(细分更细)。常用0.4-0.8 -
algorithm:也可以试试Leiden,通常更稳定
聚类结果写入:pbmc@meta.data$seurat_clusters、pbmc@active.ident
非线性降维与可视化
RunUMAP或RunTSNE:把(10–50维的)PC空间压到2维,方便可视化,同时尽量把“相似的细胞堆在一起”
- 现在更常用UMAP
pbmc4 <- RunUMAP(
pbmc4,
dims = 1:10 # 和FindNeighbors一致
)
DimPlot:展示聚类结果
DimPlot(
pbmc4,
reduction = "umap", # 用哪个降维结果("umap"/"tsne"/"pca")
label = TRUE # 在图上直接写cluster号,方便阅读
)
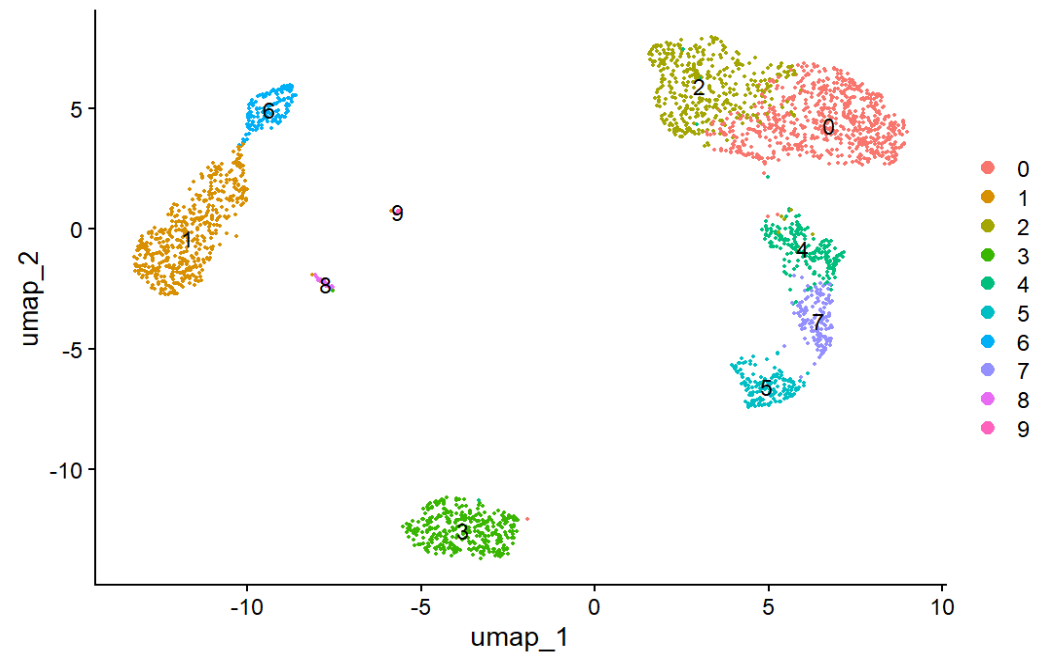
找marker基因 & 细胞类型注释
FindAllMarkers:每个cluster对其他所有细胞
- 对每个cluster与其他细胞做差异表达,找“在本cluster高表达、在别处低”的标志基因
pbmc4.markers <- FindAllMarkers(
pbmc4,
only.pos = TRUE, # 只保留高表达的marker(不关心在本cluster低表达的基因)
min.pct = 0.25, # 这个基因在某cluster里至少要在25%的细胞中表达
logfc.threshold = 0.25, # logFC至少0.25
test.use = "wilcox" # Wilcoxon秩和检验(scRNA里很常用的非参数方法)
)
head(pbmc4.markers)
pbmc4.markers %>% # 每个cluster的前几个marker
dplyr::group_by(cluster) %>%
dplyr::top_n(n = 5, wt = avg_log2FC)
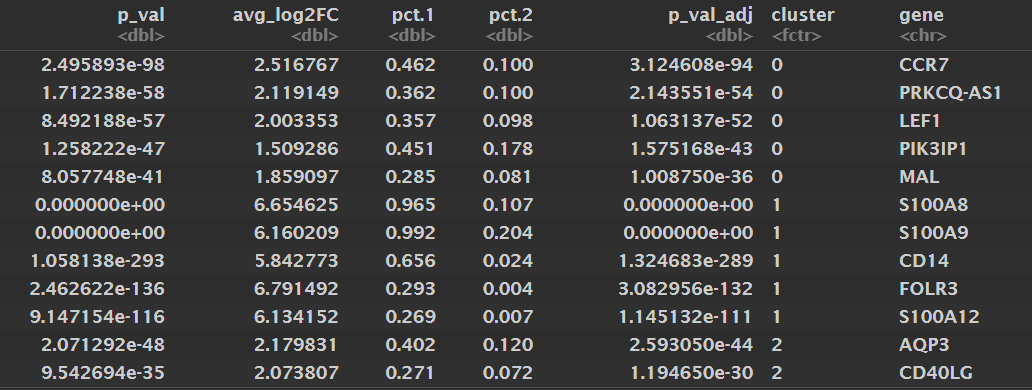
-
gene:基因名 -
cluster:这是哪个cluster的marker -
avg_log2FC:差异倍数 -
pct.1:在本cluster中表达细胞比例 -
pct.2:在其他细胞中表达比例 -
p_val_adj:多重校正后的p值
常见可视化:VlnPlot/FeaturePlot/DotPlot/DoHeatmap
如果我怀疑cluster 0是T细胞,又知道T细胞有一些标志基因(CD3D/CD3E)
FeaturePlot(pbmc4, features = c("CD3D", "CD3E"))
VlnPlot(pbmc4, features = c("CD3D"))
DotPlot(pbmc4, features = c("CD3D", "MS4A1", "LYZ", "GNLY")) + RotatedAxis()
-
FeaturePlot:在UMAP上画“这个基因在哪些细胞高表达”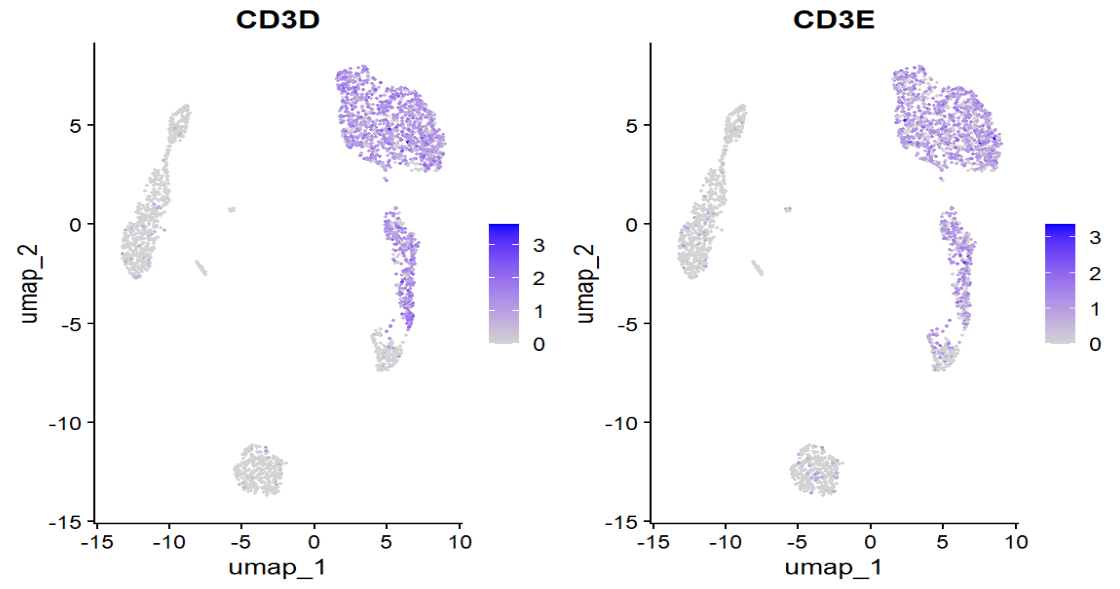
-
VlnPlot:按cluster看表达分布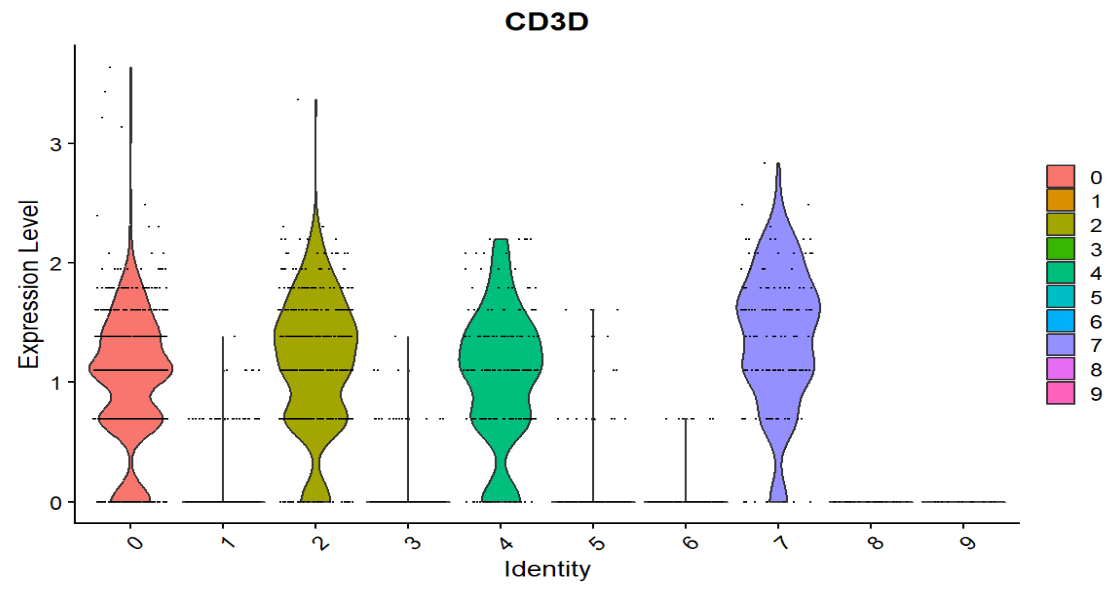
-
DotPlot:同时看多基因-多cluster: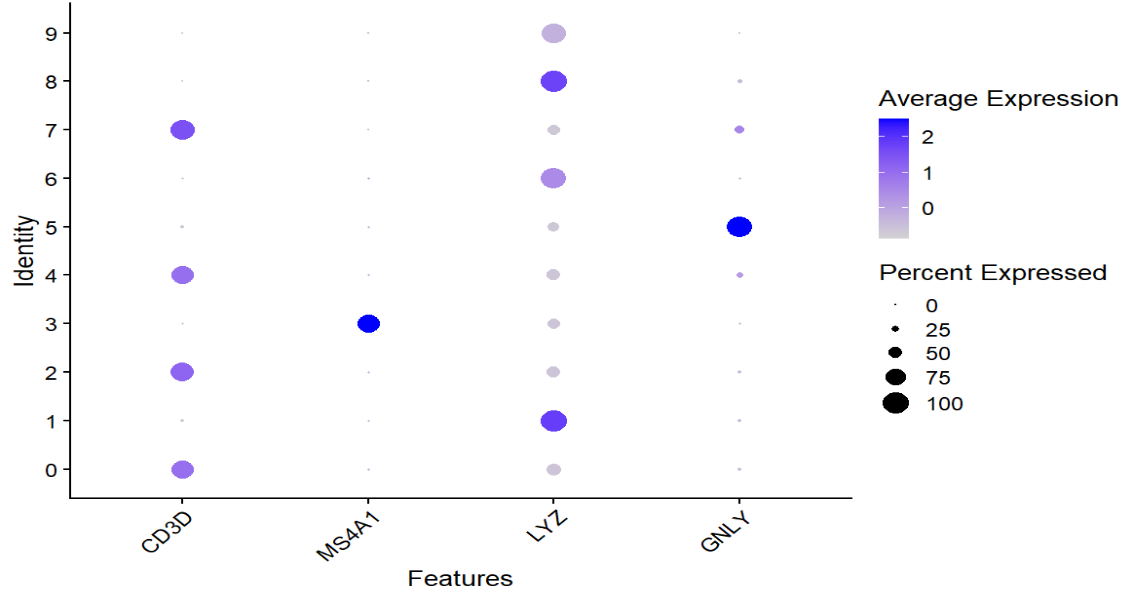
- 点的大小 = 表达细胞比例
- 颜色深浅 = 平均表达量
做一个热图,看每个cluster的top markers
top10 <- pbmc4.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC)
DoHeatmap(pbmc4, features = top10$gene) + NoLegend()
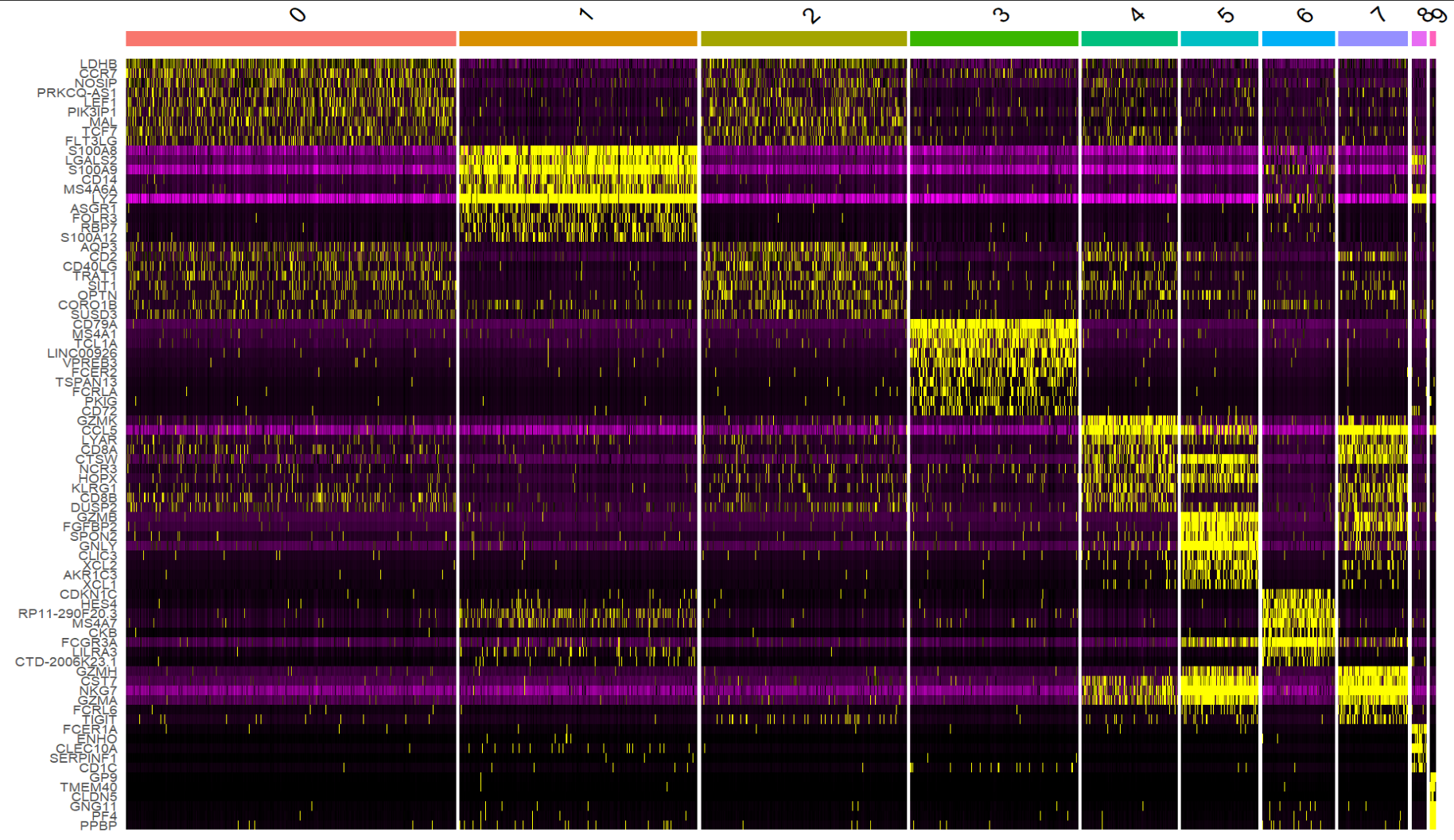
手动注释细胞类型:给cluster命名
new.cluster.ids <- c(
"Naive CD4 T", "Memory CD4 T", "CD14+ Mono",
"B cell", "CD8 T", "NK", "DC", "Megakaryocytes",
"cell 9", "cell 10"
)
names(new.cluster.ids) <- levels(pbmc4)
pbmc4 <- RenameIdents(pbmc4, new.cluster.ids) # 重命名
DimPlot(pbmc4, reduction = "umap", label = TRUE)
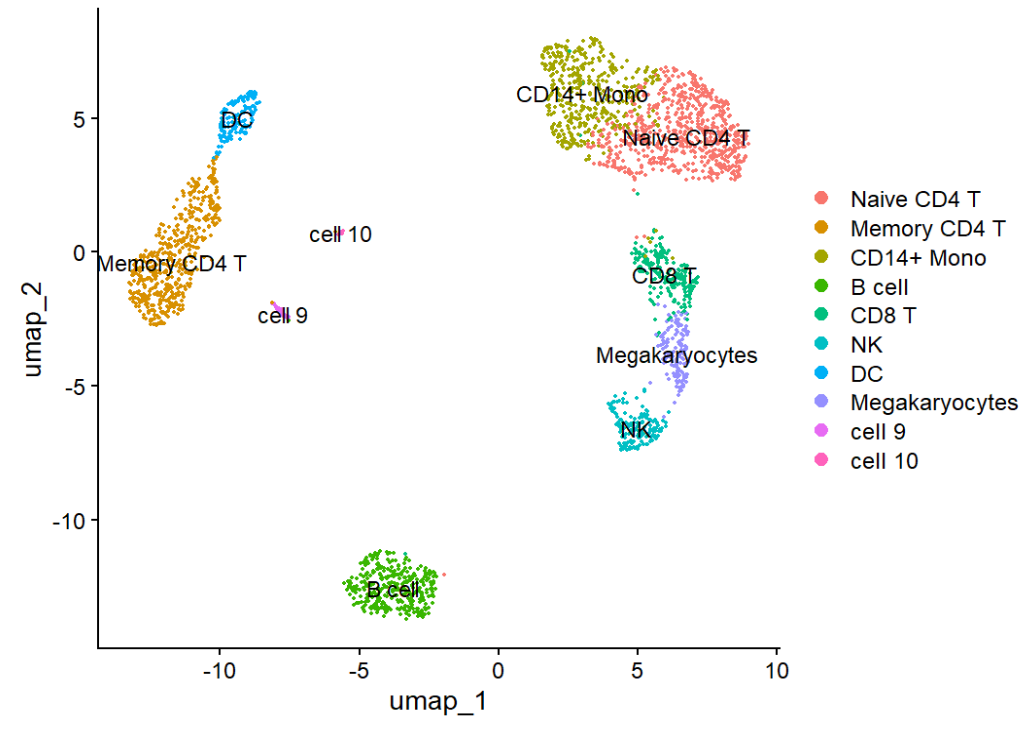
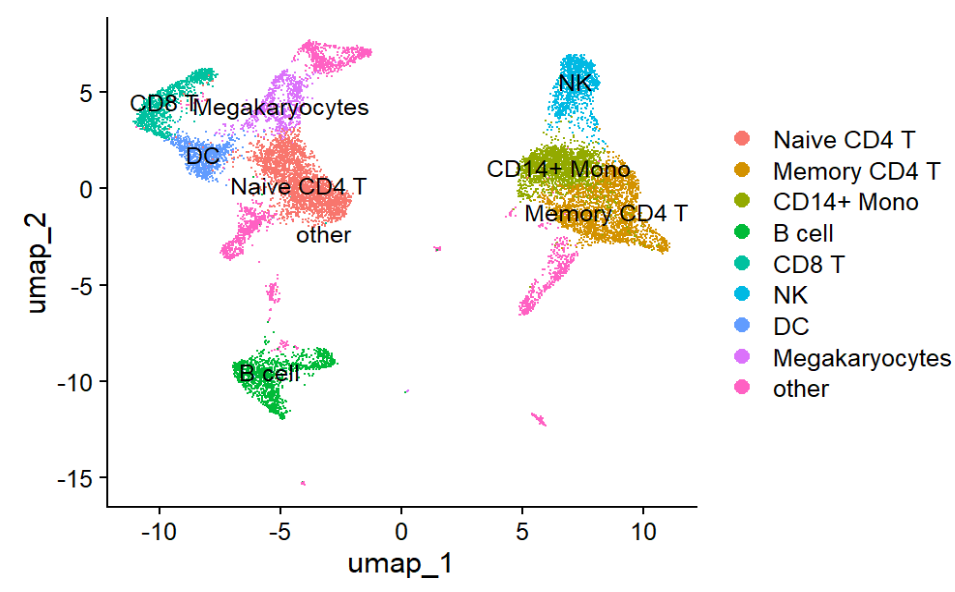
ifnb数据的分析
载入数据
library(ifnb.SeuratData)
data("ifnb")
ifnb <- UpdateSeuratObject(object = ifnb)
unique(ifnb@meta.data$stim)
可以看到有两个样本——CTRL/STIM
- CTRL:未刺激样本
- STIM:IFN-β刺激样本
按样本拆分对象 & 各自做基础预处理
先把Stim和CTRL拆开,各自做Normalize/HVG/Scale/PCA。后面再把它们整合到同一个低维空间,去掉batch/条件带来的技术差异
ifnb.list <- SplitObject(ifnb, split.by = "stim")
# 预处理
for (i in 1:length(ifnb.list)) {
ifnb.list[[i]] <- NormalizeData(ifnb.list[[i]])
ifnb.list[[i]] <- FindVariableFeatures(ifnb.list[[i]], selection.method = "vst", nfeatures = 2000)
ifnb.list[[i]] <- ScaleData(ifnb.list[[i]])
ifnb.list[[i]] <- RunPCA(ifnb.list[[i]], features = VariableFeatures(ifnb.list[[i]]))
}
- 为什么没做质控:这套数据已经做过过滤了。如果是一般数据,也是先拆开再分别质控
-
为什么要先拆开再预处理:
- HVG要在各自样本内部选:如果混在一起选,容易被样本间差异/文库大小差异主导。例如某个基因如果只在STIM里表达,就会被认为是高变,但实际上是测序差异而不是细胞类型差异
在每个样本中单独找HVG,后面再找“在至少两个样本中都变异较大”的基因,这样更合理
- 各自PCA,避免一个大样本完全主导方向:如果把把所有细胞拼一起PCA,大样本会压制小样本的结构,主成分很容易全是大样本内部的变化。分开后能平衡各样本的贡献
使用锚点做数据整合
找整合锚点(anchors):FindIntegrationAnchors
- 在两个数据集里找到“相互对应”的细胞对,把这些细胞当成“桥”,来学习两者之间的转换关系
为什么要有这步:我希望看到“同一种细胞类型在两个样本中能对齐到一起”,而且在UMAP上不会因为批次/技术导致“同一细胞类型被分到两个完全不同的块”
- 在不整合的情况下,同一细胞类型可能会被分到两堆,一堆全是CTRL,一堆全是STIM
- 整合后,两堆混成了一堆,可以在里面直接比较Stim-Ctrl的表达变化
换句话说,需要找到一个对应关系——哪个STIM细胞,看起来就是某个CTRL细胞在另一个batch/条件下的版本
# 选一批在两边都重要的基因用来对齐(默认3000个)
features <- SelectIntegrationFeatures(object.list = ifnb.list, nfeatures = 3000)
# 找锚点(锚定最近邻+校正)
ifnb.anchors <- FindIntegrationAnchors(
object.list = ifnb.list,
anchor.features = features,
reduction = "rpca" # 推荐,速度快,对大数据也友好
)
anchors:可以把它想成“跨样本的互相最近邻”。先在每个样本内部做PCA,得到两个样本各自的PC空间;再把两个样本的PCA空间对齐坐标系(reduction = "rpca"),即先对所有样本做一次“参考PCA”,再把各自的 PC 表达重新投到这个共享坐标系;在共享低维空间里,寻找互相最近的细胞对(对于每个CTRL细胞,在STIM中找K个最近邻),那些“你是我的最近邻,我也是你的最近邻”的细胞对,就是候选anchor;对候选anchors打分过滤(看它们的局部环境是否也相似)
- 最终,每个anchor大致包含“来自样本A的某个细胞i,来自样本B的某个细胞j”,以及一些权重/分数(标明可信度)
- 简单的说,一个anchor就是“认为这两个细胞本质上是同一个细胞类型/类似状态,只是技术和条件不同”
整合数据:IntegrateData
- 整合后的表达存到一个新的assay——
integrated - 后面用于PCA/UMAP/聚类的都是这个integrated assay
ifnb.integrated <- IntegrateData(anchorset = ifnb.anchors)
DefaultAssay(ifnb.integrated) <- "integrated"
ifnb.integrated
利用这些anchor来学习一个“从B样本映射到A样本”的校正函数,然后把所有B细胞都往这个方向拉,让两边的相同细胞类型对齐
在整合后的空间里做PCA/UMAP/聚类(同前)
ifnb.integrated <- ScaleData(ifnb.integrated, verbose = FALSE)
ifnb.integrated <- RunPCA(ifnb.integrated, npcs = 30, verbose = FALSE)
ifnb.integrated <- RunUMAP(ifnb.integrated, reduction = "pca", dims = 1:30)
ifnb.integrated <- FindNeighbors(ifnb.integrated, reduction = "pca", dims = 1:30)
ifnb.integrated <- FindClusters(ifnb.integrated, resolution = 0.4)
两个最重要的图:
# 看cluster分布
DimPlot(ifnb.integrated, reduction = "umap", label = TRUE)
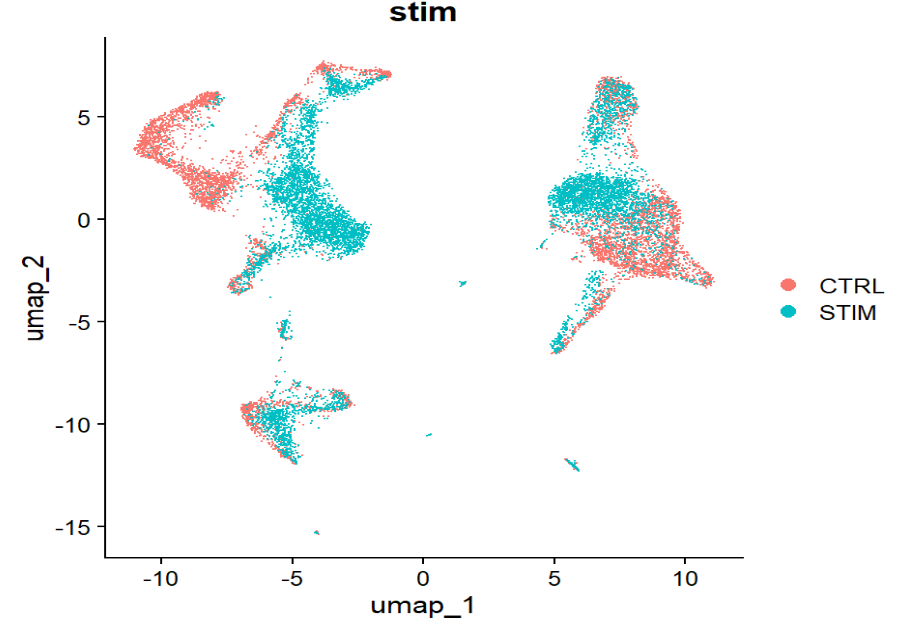
# 看Stim/CTRL在同一UMAP上的混合程度
DimPlot(ifnb.integrated, reduction = "umap", group.by = "stim")
理想情况下:
- cluster分布:每个cluster形状比较清晰
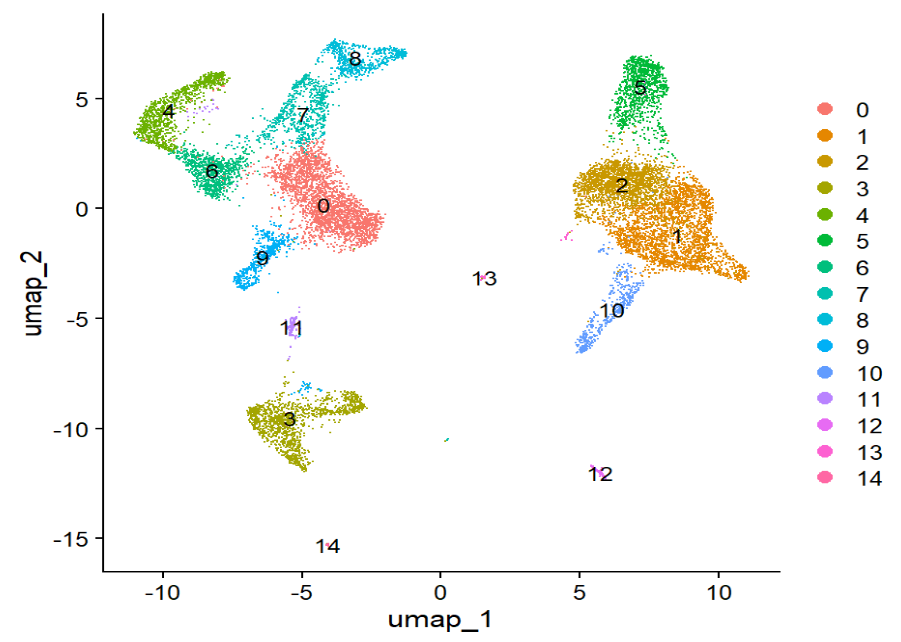
- 混合程度:大多数cluster里Stim和CTRL都有,而不是“某个cluster全是Stim,另一个全是CTRL”。这说明整合把batch/条件的技术差异对齐了,而保留了真正的细胞类型结构
在整合后空间里做细胞类型注释
找marker & 按cluster注释(同前)
DefaultAssay(ifnb.integrated) <- "RNA" # 找marker建议用原始RNA,而不是integrated
ifnb.markers <- FindAllMarkers(
ifnb.integrated,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.25
)
# 每个cluster取前5个marker
top5 <- ifnb.markers %>%
group_by(cluster) %>%
top_n(5, wt = avg_log2FC)
top5
# 以免疫相关标志基因为例
FeaturePlot(ifnb.integrated, features = c("MS4A1", "CD79A")) # B cells
FeaturePlot(ifnb.integrated, features = c("CD3D", "CD4", "CCR7")) # CD4 T
FeaturePlot(ifnb.integrated, features = c("NKG7", "GNLY")) # NK / CD8 cytotoxic
FeaturePlot(ifnb.integrated, features = c("LYZ")) # Monocytes
# 给 cluster 起名字
new.cluster.ids <- c(
"Naive CD4 T", "Memory CD4 T", "CD14+ Mono", "B cell",
"CD8 T", "NK", "DC", "Megakaryocytes", rep("other", 7)
)
# RenameIdents(很重要),如果不写后面分组时会报错
names(new.cluster.ids) <- levels(ifnb.integrated)
ifnb.integrated <- RenameIdents(ifnb.integrated, new.cluster.ids)
DimPlot(ifnb.integrated, reduction = "umap", label = TRUE)
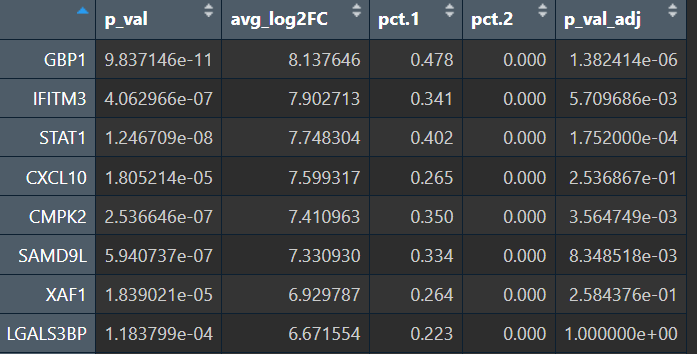
在同一细胞类型中比较Stim-CTRL的差异表达
ifnb数据的核心问题是:同一种细胞类型在IFN-β刺激前后,哪些基因/通路被激活
Seurat内置 + 细胞级DE
先只看”Naive CD4 T”这个细胞类型
# 选出Naive CD4 T的细胞
naive.cd4 <- subset(ifnb.integrated, idents = "Naive CD4 T")
# 把stim列当作分组变量
Idents(naive.cd4) <- naive.cd4$stim
# CTRL vs STIM
naive.cd4.DE <- FindMarkers(
naive.cd4,
ident.1 = "STIM",
ident.2 = "CTRL",
logfc.threshold = 0.25,
min.pct = 0.1
)
View(naive.cd4.DE)
View(naive.cd4.DE %>% # 刺激后上调最明显的基因
arrange(desc(avg_log2FC)) %>%
head(20))
pseudobulk:按照病人/样本ID,先把同一种细胞类型的表达加总成pseudobulk再用edgeR/DESeq2
- 对每个(样本 × 细胞类型)组合,把细胞counts加总
- 得到一个pseudo-bulk矩阵:基因为行,“样本 × 细胞类型”为列
- 用edgeR/DESeq2做DE
一个小问题——既然要比较Stim-CTRL,为什么还要anchor+Integrate,而不是直接拿“原始数据”比?
- 注意anchor+Integrate是为了了解不同样本间的技术偏移,消除批次效应
- 在真正做差异表达时,我们实际上是先在integrated空间里聚类 & 注释,得到“Naive CD4 T”这一个群,再把这个群的细胞按Stim/Ctrl分两个组,比较同一细胞类型内两个样本的表达差别,这时使用的是原始表达(RNA assay),不使用integrated里的表达值
- 因为它已经被校正了,可能存在“整合时把’某个基因在Stim整体表达偏高’当成批次效应来校正”
- 同时
FindMarkers(无论使用 Wilcoxon/t-test/MAST哪种方法)都隐含假设:输入的是某种“真实的表达测量”——log-normalized counts或近似(对应RNA assay中的@counts-原始UMI count或@data-log-normalized处理),复杂校正后的值的p值和logFC就不那么严谨了
- 如果两个样本的批次效应很小,其实可以也不整合,但通常批次效应都很强,不整合的话容易让cluster按照样本来源而不是细胞类型在UMAP上分块
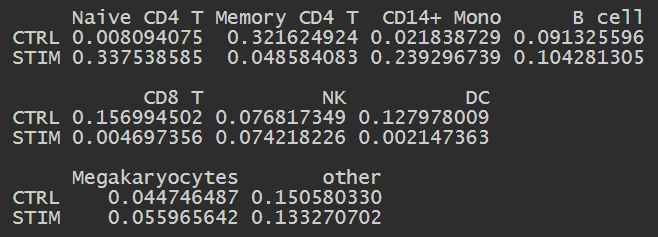
看细胞组成变化:Stim-CTRL中各细胞类型比例
这也是整合的好处之一:可以在同一空间里看Stim前后细胞类型的比例变化
用prop.table快速看比例:
tab <- table(ifnb.integrated$stim, Idents(ifnb.integrated)) # 取出meta.data里的stim和cell type
prop.table(tab, margin = 1) # 按行(每个条件)归一化
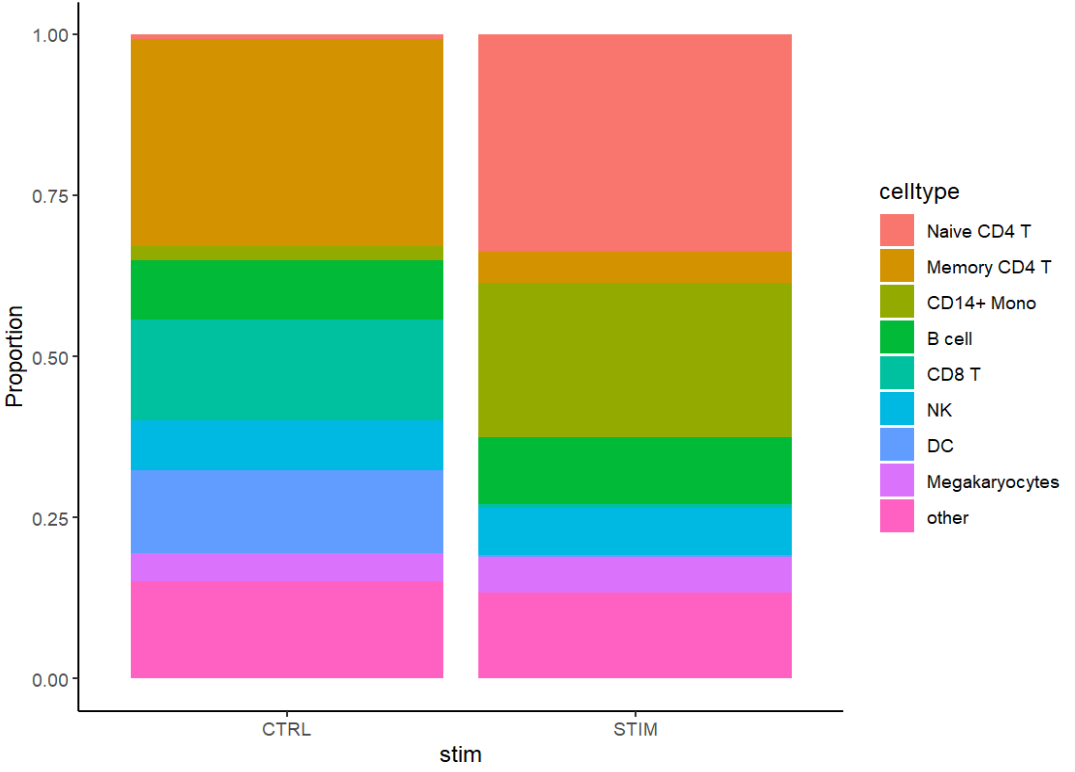
用ggplot做一个简单的堆叠条形图:
# 注意是不是严格的统计检验,只是直观可视化
df.comp <- as.data.frame(tab)
colnames(df.comp) <- c("stim", "celltype", "count")
ggplot(df.comp, aes(x = stim, y = count, fill = celltype)) +
geom_bar(stat = "identity", position = "fill") +
ylab("Proportion") +
theme_classic()
总结:
- 对每个样本独立做QC/预处理/PCA
- 用anchors+IntegrateData在一个共同空间里对齐细胞类型,做聚类和可视化
- 一旦celltype/cluster定好了,就回到RNA assay(或原始counts)上
- 如果要做更严谨的DE(尤其多病人、多样本),进一步用pseudobulk+edgeR/DESeq2在样本层面建模
使用SCTransform整合工作流
是Seurat官方推荐的方法,SCTransform对技术噪声建模更合理,整合效果通常比LogNormalize稳定
# 1. 拆分对象
ifnb.list <- SplitObject(ifnb, split.by = "stim")
# 2. 每个对象分别SCTransform
for (i in 1:length(ifnb.list)) {
ifnb.list[[i]] <- SCTransform(ifnb.list[[i]], verbose = FALSE)
}
# 3. 选整合特征 + 准备整合
features <- SelectIntegrationFeatures(ifnb.list, nfeatures = 3000)
ifnb.list <- PrepSCTIntegration(object.list = ifnb.list, anchor.features = features)
# 4. 找anchors & 整合
ifnb.anchors <- FindIntegrationAnchors(
object.list = ifnb.list,
normalization.method = "SCT",
anchor.features = features
)
ifnb.integrated.sct <- IntegrateData(
anchorset = ifnb.anchors,
normalization.method = "SCT"
)
# 5. 后面PCA/UMAP/聚类/DE的流程基本相同(此处省略部分)
ifnb.integrated.sct <- ScaleData(ifnb.integrated.sct, verbose = FALSE)
ifnb.integrated.sct <- RunPCA(ifnb.integrated.sct, npcs = 30, verbose = FALSE)
ifnb.integrated.sct <- RunUMAP(ifnb.integrated.sct, reduction = "pca", dims = 1:30)
ifnb.integrated.sct <- FindNeighbors(ifnb.integrated.sct, reduction = "pca", dims = 1:30)
ifnb.integrated.sct <- FindClusters(ifnb.integrated.sct, resolution = 0.4)
DimPlot(ifnb.integrated.sct, reduction = "umap", label = TRUE)
DimPlot(ifnb.integrated.sct, reduction = "umap", group.by = "stim")
GSE233208
论文对snRNA数据的处理流程:QC→整合(Harmony/scVI)→聚类→注释
- 看细胞状态abundance随疾病变化(MiloR)
- 各celltype/cellstate内做疾病vs对照DE(用MAST模型,带协变量)
- 构建共表达网络(hdWGCNA),找模块(M1、M11等),看在不同疾病组/脑区里的变化
- 用scDRS把ADGWAS基因集的信号映射到单细胞,看看哪些celltype/state富集AD遗传风险
- 把snRNA的cellstates映射到Visium空间里(CellTrek),看这些cellstate/模块在大脑皮层层次、白质、斑块周围的分布
- 再往上做cell–cellcommunication(CellChat)、amyloidhotspot分析等
- 最后建立一个大规模人类+小鼠的snRNA+空间转录组图谱(Control/EarlyAD/LateAD/DSAD等)
单细胞分析部分
GEO中的数据:人类snRNA-seq总共有≈58万个nuclei,主要细胞类型包括EX(兴奋性神经元)、INH(抑制性神经元)、MG(小胶质)、ASC(星形胶质)、ODC/OPC(少突及前体)、内皮、周细胞、成纤维细胞等
- 这里先使用
GSE233208_Human_snRNA-Seq_ADDS_integrated.rds.gz:人类snRNA-seq,已经整合好的Seurat对象
library(Seurat)
library(tidyverse)
library(ggpubr)
# BiocManager::install("MAST")
library(MAST)
library(clusterProfiler)
library(org.Hs.eg.db)
# 注:因为原rds很大,这里提取了大概1/4的细胞做试分析
sn <- readRDS("C:\\Users\\17185\\Desktop\\hERV_calc\\GSE233208\\data\\GSE233208_human_snRNA_subset.rds")
DefaultAssay(sn) # "RNA"
colnames(sn@meta.data)
table(sn$Diagnosis)
table(sn$cell_type)
table(sn$SampleID)
因为rds文件名里有一个”integrated”,这说明作者已经把多个样本(多个病人、多个脑区、多个疾病组)使用他自己的pipeline(scVI/Harmony等)做过批次/样本整合,所以就不需要整合这步,只需基于这个整合好的对象做一些下游分析
预处理:同前
sn <- NormalizeData(sn)
sn <- FindVariableFeatures(sn)
sn <- ScaleData(sn)
sn <- RunPCA(sn)
sn <- FindNeighbors(sn, dims = 1:30)
sn <- FindClusters(sn, resolution = 0.4)
sn <- RunUMAP(sn, dims = 1:30)
注:这里的Normalize/PCA/UMAP,是在同一个统一空间里微调,而不是在“多个完全独立的原始样本之间”第一次对齐
初步可视化:看细胞类型和疾病在UMAP上的分布
# 按聚类看
DimPlot(sn, reduction = "umap", label = TRUE)
# 按细胞类型看
DimPlot(sn, reduction = "umap", group.by = "cell_type", label = TRUE)
# 按有无AD看(Diagnosis:Control/DSAD)
DimPlot(sn, reduction = "umap", group.by = "Diagnosis")
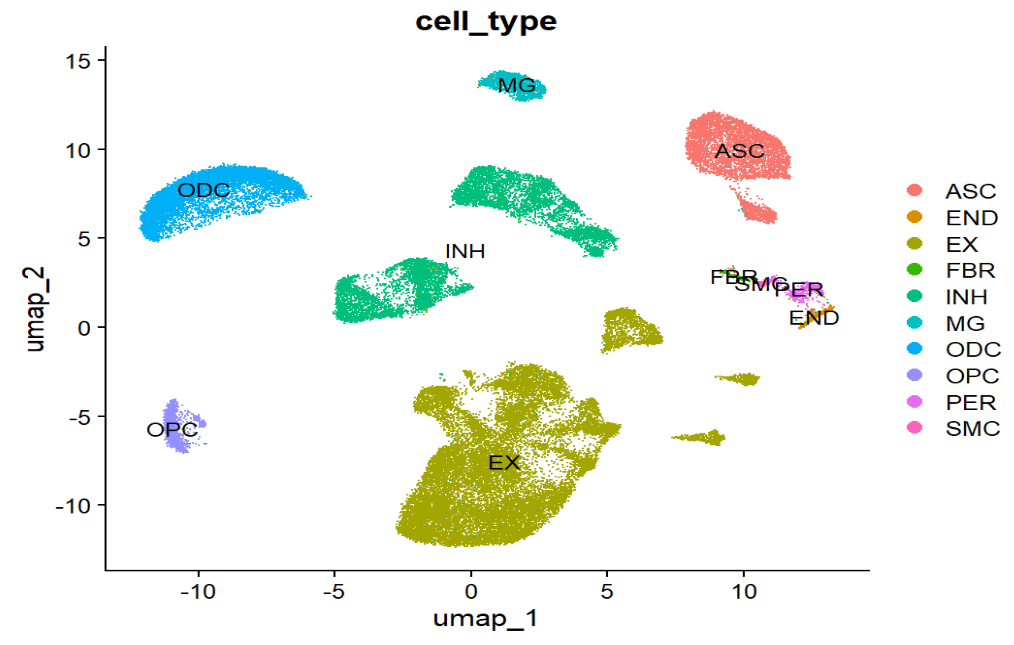
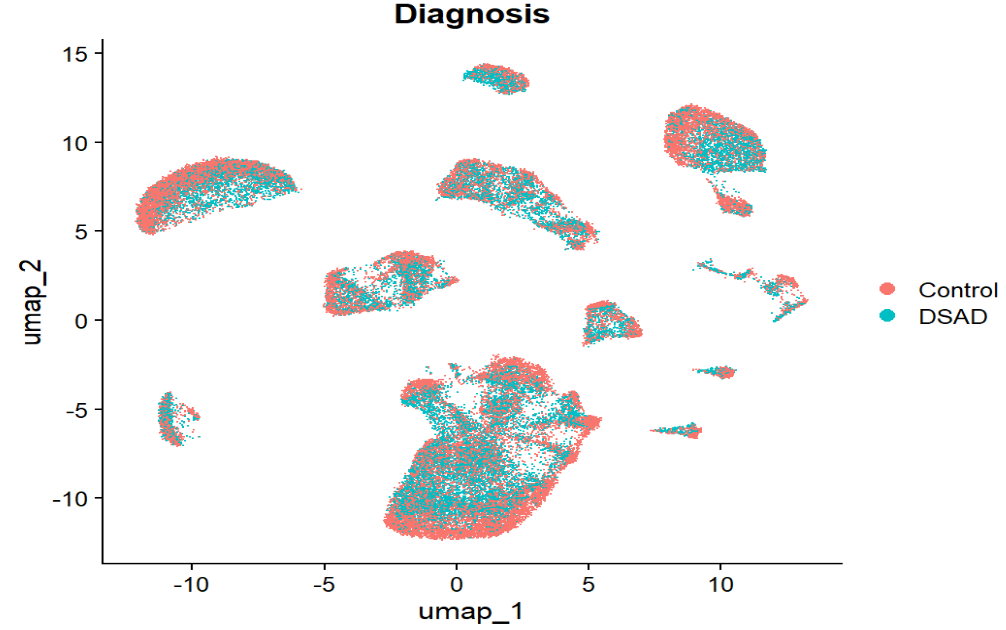
看不同大类细胞类型的位置(EX、INH、MG、ASC、ODC等),再看Diagnosis上色,大致观察:
- 是否有某些cluster主要来自DSAD(提示cell state变化)
- 或者Control/DSAD均匀分布(提示更像“每种细胞类型里表达变化”,而不是“出现完全新的细胞类型”)
这里看起来像是均匀分布的,并没有某种细胞类型都是DSAD组的
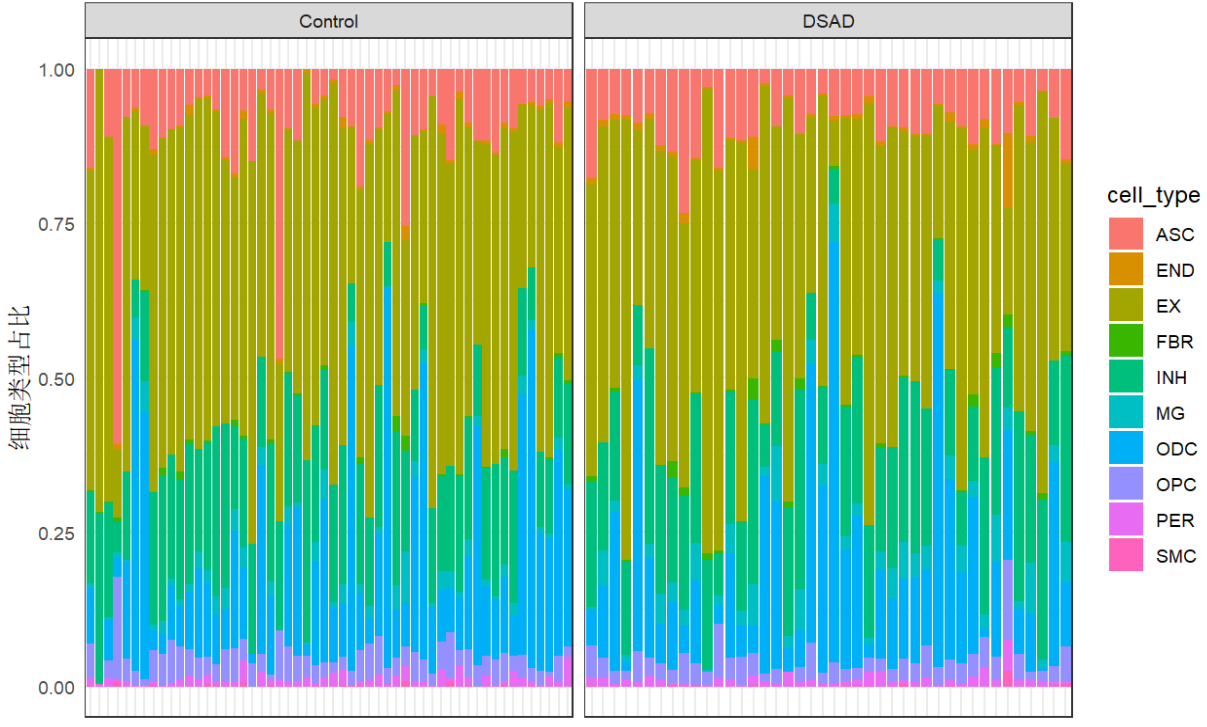
比较Control vs DSAD的细胞组成
这里直接ggplot画图了,原文中使用了MiloR做统计检验
# 统计每个样本里各细胞类型的比例
cell_comp <- sn@meta.data %>%
group_by(SampleID, Diagnosis, cell_type) %>%
summarise(n = n(), .groups = "drop") %>%
group_by(SampleID) %>%
mutate(freq = n / sum(n))
head(cell_comp)
# 堆叠条形图:每个样本的细胞类型组成
ggplot(cell_comp, aes(x = SampleID, y = freq, fill = cell_type)) +
geom_bar(stat = "identity") +
facet_wrap(~ Diagnosis, scales = "free_x") +
ylab("细胞类型占比") +
xlab("") +
theme_bw() +
theme(axis.text.x = element_blank(), axis.ticks=element_blank())
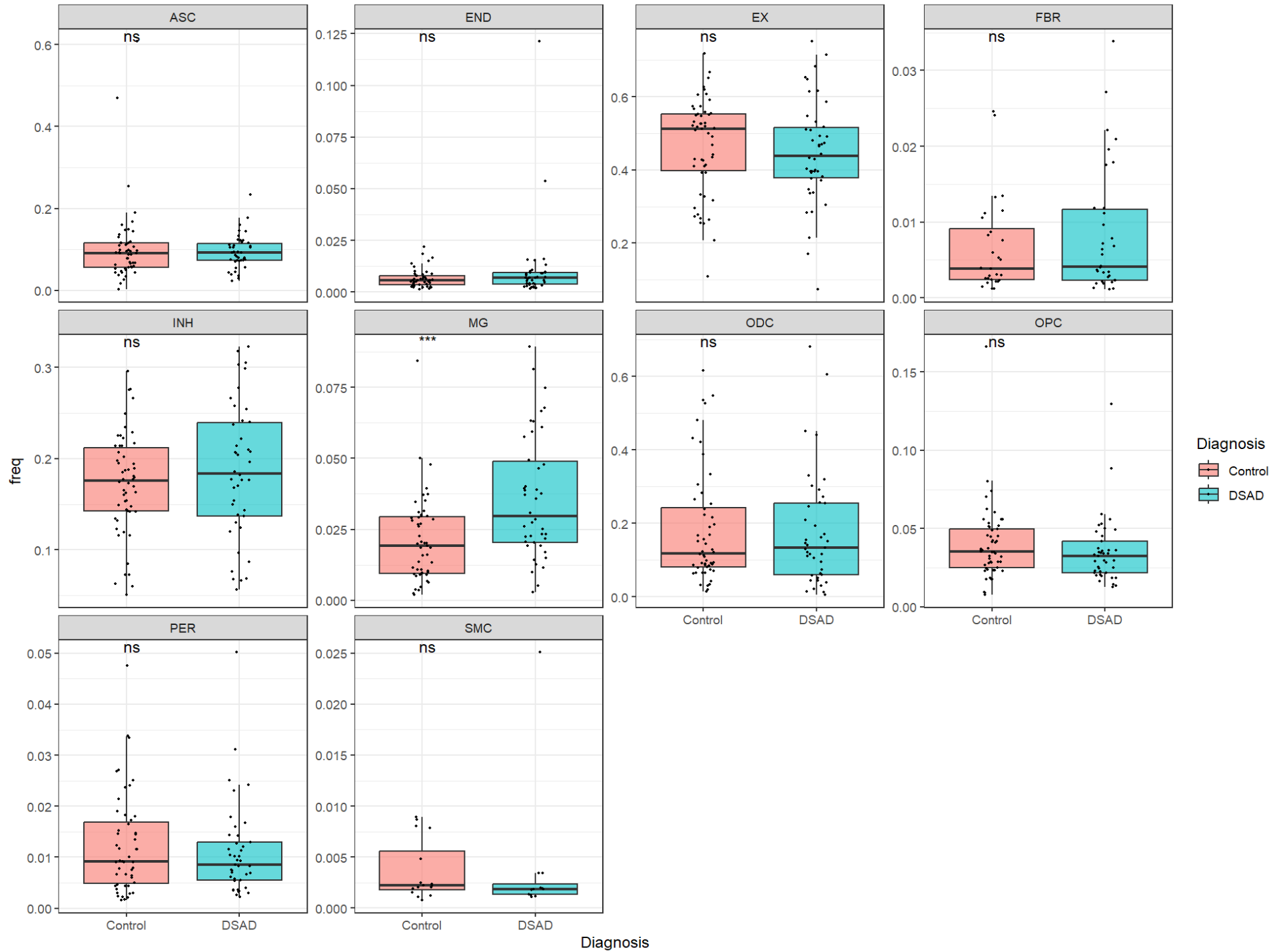
# 箱型图:每个细胞都画一张图,标识在两个样本中的占比
ggplot(cell_comp, aes(x = Diagnosis, y = freq, fill = Diagnosis)) +
geom_boxplot(outlier.shape = NA, alpha = 0.6) +
geom_jitter(width = 0.1, size = 0.5) +
facet_wrap(~ cell_type, scales = "free_y") +
theme_bw() +
stat_compare_means(
method = "wilcox.test",
label = "p.signif"
)
在某个细胞类型内做DSAD vs Control的差异表达
原文使用了MAST GLM模型,协变量包括测序批次、每个细胞的UMI数、PMI、性别等
这里使用FindMarkers(test.use = "MAST")做一个简化版,根据上面粗略的检验结果,microglia(MG)这个细胞类型在两个样本中占比有显著不同,可能存在差异基因最多
Idents(sn) <- "cell_type" # 按细胞类型设定身份
mg <- subset(sn, idents = "MG") # 取出MG细胞
Idents(mg) <- mg$Diagnosis # mg中Diagnosis作分组变量
mg_DE <- FindMarkers(
mg,
ident.1 = "DSAD",
ident.2 = "Control", # DSAD相对Control的变化
test.use = "MAST", # 使用MAST模型
logfc.threshold = 0.25,
min.pct = 0.1,
latent.vars = c("nCount_RNA", "Batch", "PMI", "Sex") # 协变量
)
mg_DE_top <- mg_DE %>%
dplyr::filter(abs(avg_log2FC)>0.5)
arrange(p_val_adj) %>%
head(20)
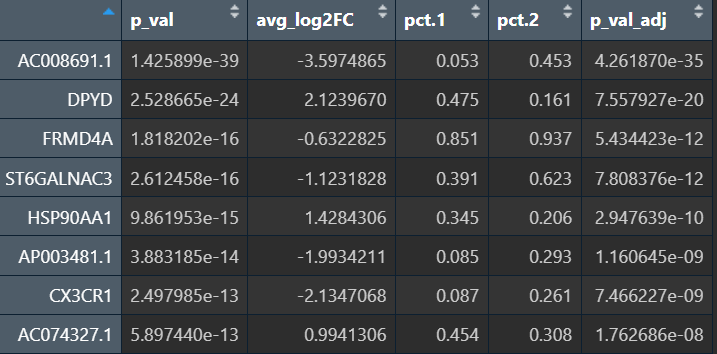
注:这里按p值排序,是想找“统计上最显著”的基因,通常是各种经典AD相关基因,便于快速对照论文结果(某个通路在这个细胞类型中最显著)
- 如果是想找生物效应最强,或者是想做一个“top up/down”的基因表就用logFC排
- 在更多的时候,是先按p和logFC设阈值(筛掉既不显著又很小的差异),再按logFC排,如下
- 在单细胞分析中,易出现“假大logFC”的情况,比如在AD组中只有5个细胞表达某基因且都很高,正常组几乎全0,这样logFC就会很大,但在整个细胞类型层面影响很小。因此
FindMarkers会设置min.pct = 0.1的过滤,要求至少10%的细胞表达
mg_DE_up <- mg_DE %>% # 取显著上调的基因
dplyr::filter(p_val_adj < 0.001, avg_log2FC > 0.5) %>%
arrange(desc(avg_log2FC)) %>%
head(20)
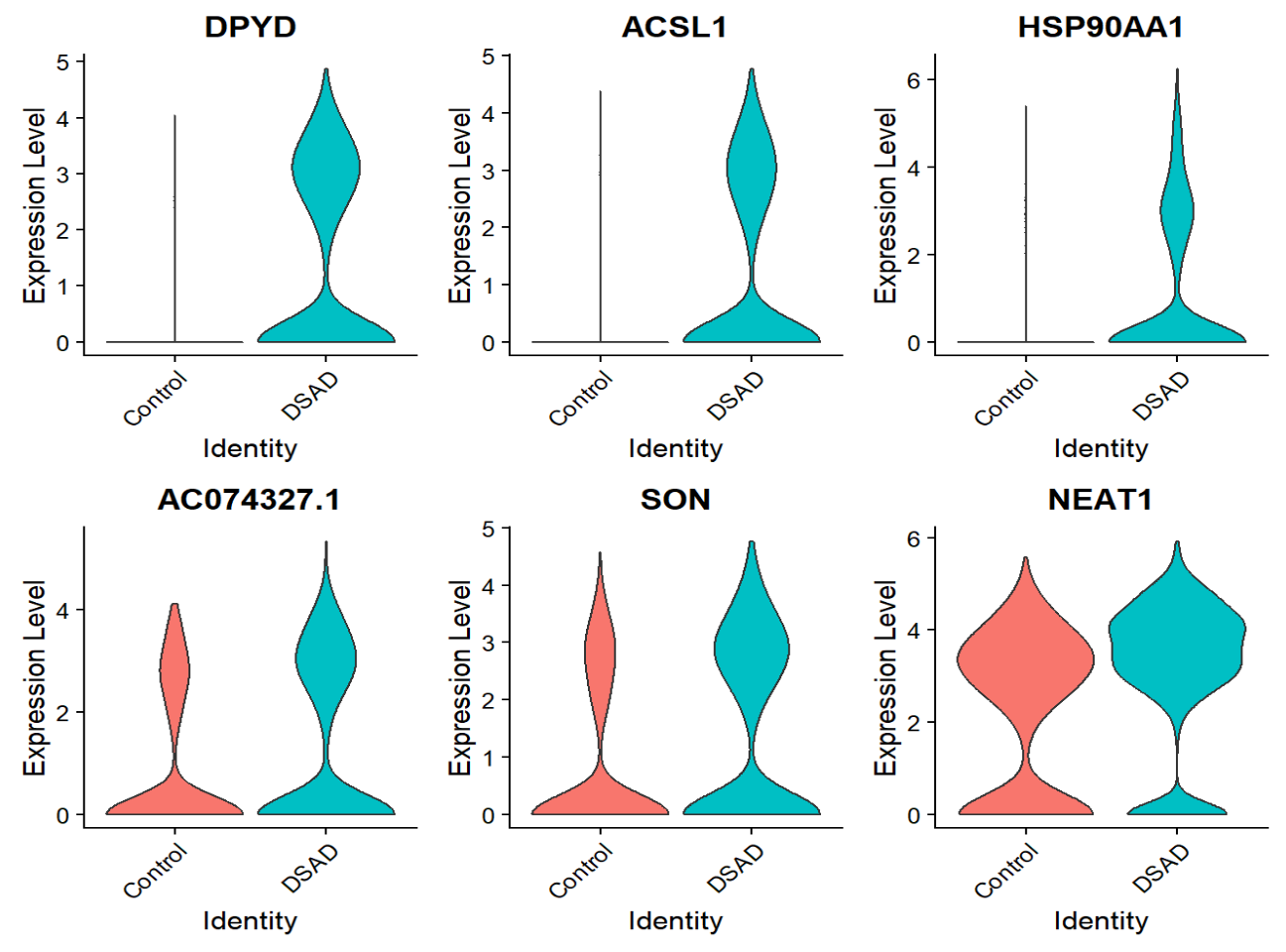
# 小提琴图:看某些基因在MG中DSAD vs Control的差异
VlnPlot(mg, features = rownames(mg_DE_up)[1:6], group.by = "Diagnosis", pt.size = 0)
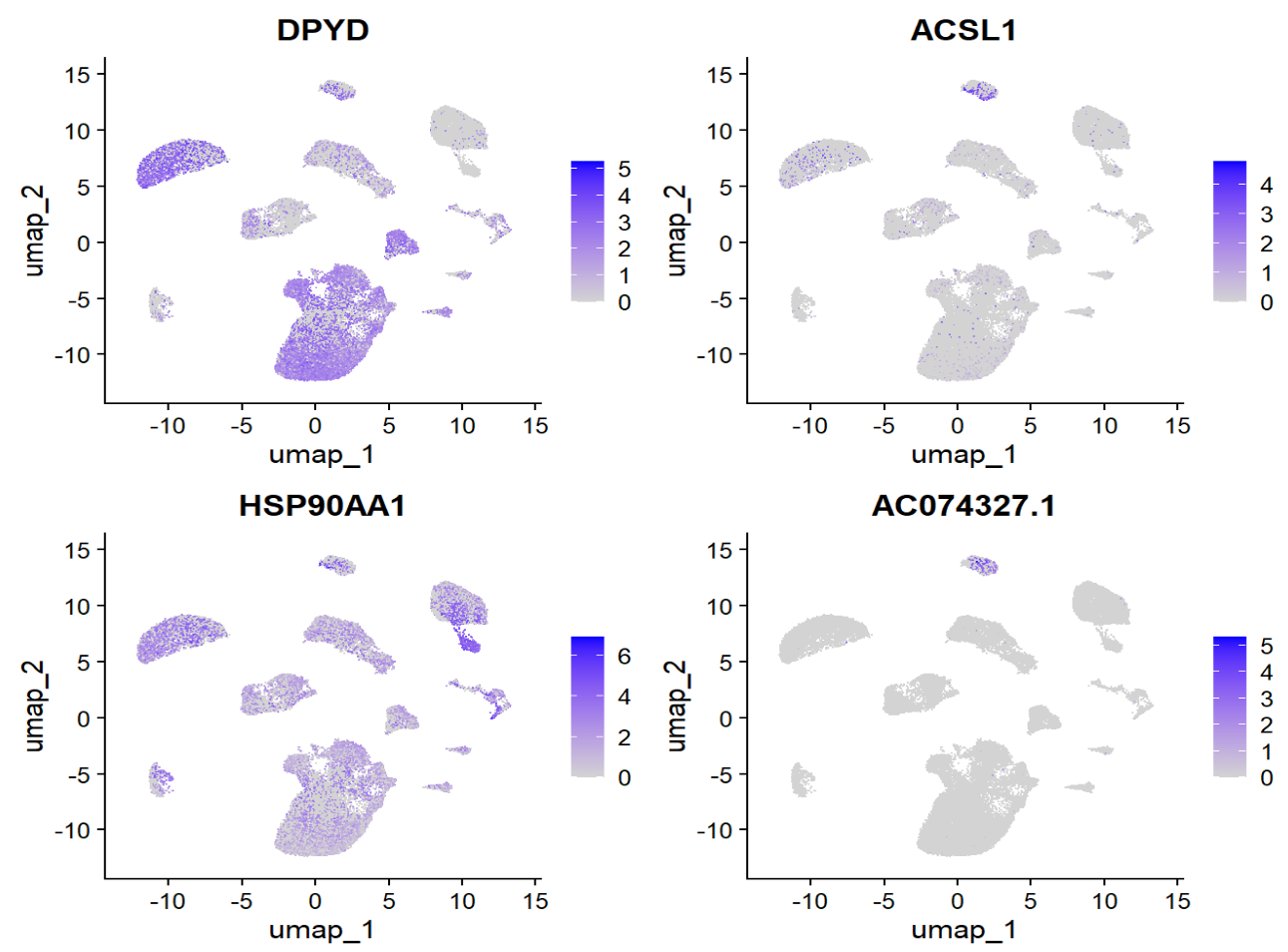
# 在UMAP上(整个数据中)看这些基因表达在哪些细胞类型强
FeaturePlot(sn, features = rownames(mg_DE_up)[1:4])
简单的GO/KEGG富集:
# 因为原来的阈值筛出来的基因太少,做GO富集结果很奇怪,这里就放宽了阈值
mg_DE2 <- FindMarkers(
mg,
ident.1 = "DSAD",
ident.2 = "Control",
test.use = "MAST",
logfc.threshold = 0.1,
min.pct = 0.1,
latent.vars = c("nCount_RNA", "Batch", "PMI", "Sex")
)
# 不按p值筛选,直接取前100个
mg_DE_up <- mg_DE2 %>%
dplyr::filter(avg_log2FC > 0) %>%
arrange(desc(p_val_adj))
mg_DE_up_gene <- as.character(rownames(mg_DE_up))[1:100]
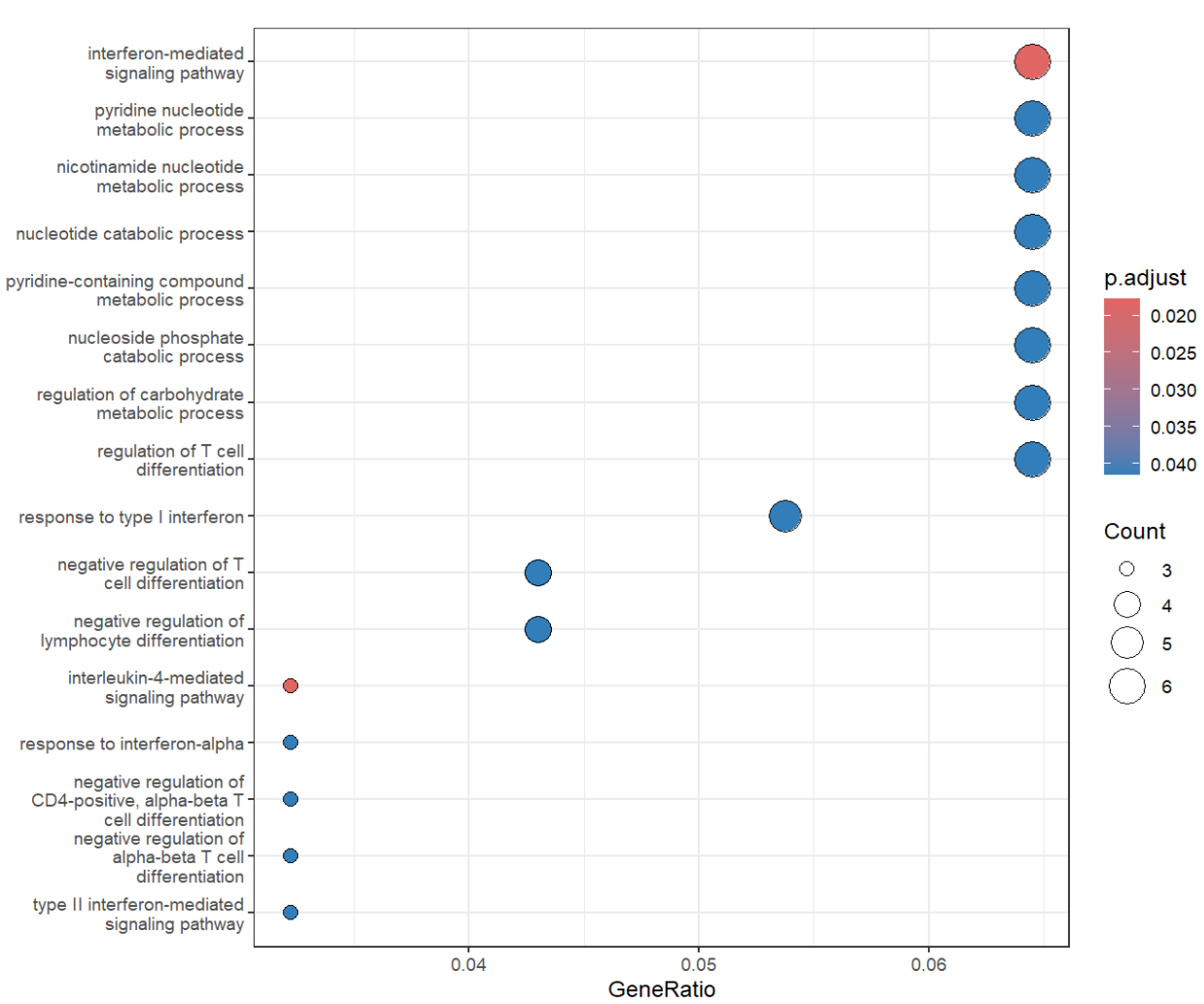
ego <- enrichGO(
gene = mg_DE_up_gene,
OrgDb = org.Hs.eg.db,
keyType = "SYMBOL",
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.2,
minGSSize = 5
)
dotplot(ego, showCategory = 20) +
theme_bw()
- 为什么常用
ont = BP:想看AD中某个细胞类型发生了哪些“生物学改变”(炎症反应、小胶质活化、髓鞘形成/损伤、血管重构、突触剪枝),这些都属于“生物学过程(BP)”的范畴。”MF”是“这些蛋白本身是什么功能”(受体、激酶……),”CC”是“这些蛋白主要在哪儿”(突触、线粒体、核膜……) - 因为是抽样的细胞,每组细胞数量较少,所以分析得到的差异基因也比较少
在细胞类型内做亚群+差异丰度
- 不仅有MG这样的大类细胞类型,还有例如MG1、MG2这样的细胞状态(cell states),作者用MiloR看这些状态再两组中是否富集
- 论文中强调:很多变化不是“多了一种完全新的细胞类型”,而是某些特定状态”的细胞比例增多或减少
- 在每个细胞类型内做子聚类 → 得到几个state → 比较每个state在Control vs DSAD中的富集情况
在某个细胞类型内做子聚类:流程同前
DefaultAssay(mg) <- "RNA"
mg <- NormalizeData(mg)
mg <- FindVariableFeatures(mg)
mg <- ScaleData(mg)
mg <- RunPCA(mg)
mg <- FindNeighbors(mg, dims = 1:20)
mg <- FindClusters(mg, resolution = 0.3)
mg <- RunUMAP(mg, dims = 1:20)
mg$MG_state <- Idents(mg)
给这些MG_state起名字:找各个state的的标志基因
mg_state_markers <- FindAllMarkers(
mg,
only.pos = TRUE,
logfc.threshold = 0.1,
min.pct = 0.1
)
# 将每个聚类的标志基因写入一个df中
mg_marker_gene_df <- data.frame()
for(i in unique(mg_state_markers$cluster)){
marker_df <- mg_state_markers %>%
dplyr::filter(cluster == i, p_val_adj < 0.05, avg_log2FC > 1) %>%
dplyr::arrange(p_val_adj)
marker_gene <- paste(rownames(marker_df), collapse = ';')
mg_marker_gene_df <- rbind(mg_marker_gene_df, c(i, marker_gene))
}
colnames(mg_marker_gene_df) <- c("cluster", "marker_gene")
write.csv(mg_marker_gene_df, 'mg_marker_gene.csv', col.names = T, row.names = F)
观察一下标志基因,经过GPT判断
- cluster 2:稳态microglia(Homeostatic MG)——在健康状态/疾病早期维持脑内稳态的MG(CX3CR1、P2RY12高表达)
- cluster 0:激活/疾病相关microglia(SPP1⁺/CD163⁺ DAM-like MG)——在AD/DSAD病灶附近被激活,负责任务包括吞噬Aβ/碎片、分泌炎症因子、重塑组织(SPP1、CD163、SLC2A3等基因高表达)
- cluster 1:神经元,不是MG state(细胞骨架/突触后致密物/突触粘附分子/电压门控Ca²⁺通道相关)
- cluster 3:T细胞,不是MG state(T cells/T+NK免疫标志基因)
Idents(mg) <- "MG_state"
mg_pure <- subset(mg, idents = c("0", "2"))
new.state.ids <- c("MG_DAM", "MG_homeo") # 激活/SPP1⁺ DAM-like MG --- 稳态P2RY12⁺ MG
names(new.state.ids) <- levels(mg_pure)
mg_pure <- RenameIdents(mg_pure, new.state.ids)
mg_pure$MG_state2 <- Idents(mg_pure)
# 按Diagnosis着色
DimPlot(mg_pure, reduction = "umap", group.by = "Diagnosis")
# 按MG_state着色
DimPlot(mg_pure, reduction = "umap", group.by = "MG_state2", label = TRUE)
# Diagnosis + MG_state交叉
DimPlot(mg_pure, reduction = "umap", split.by = "Diagnosis", group.by = "MG_state2")
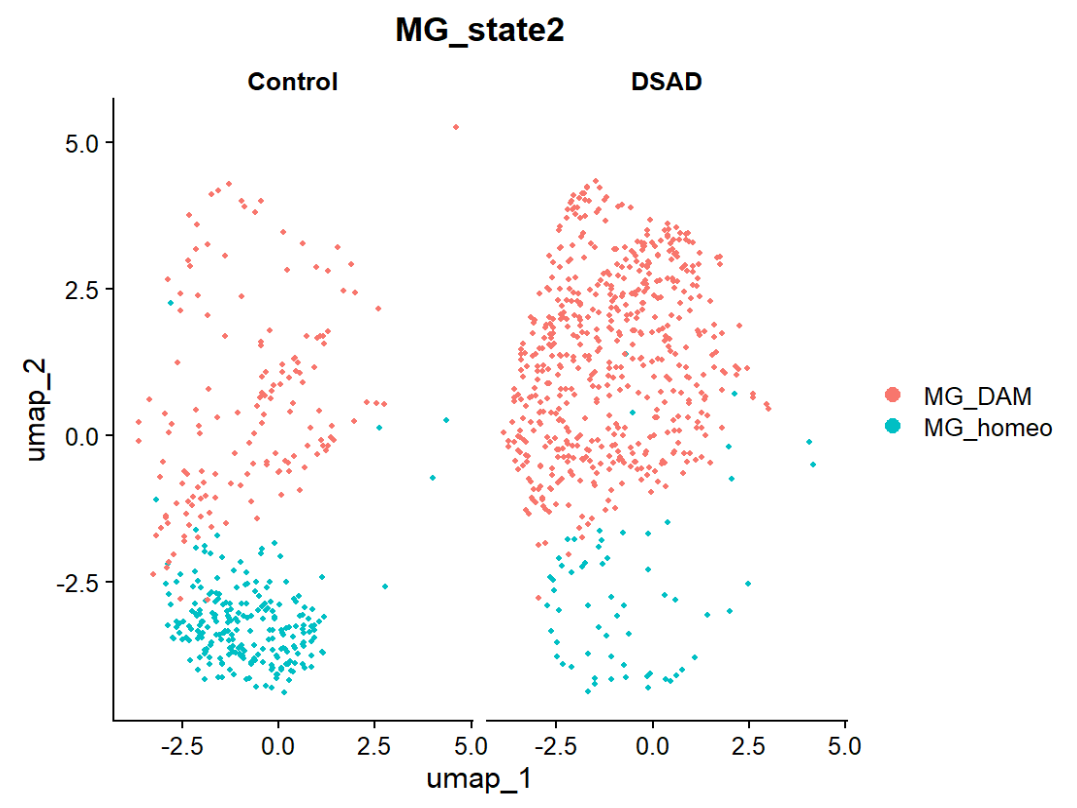
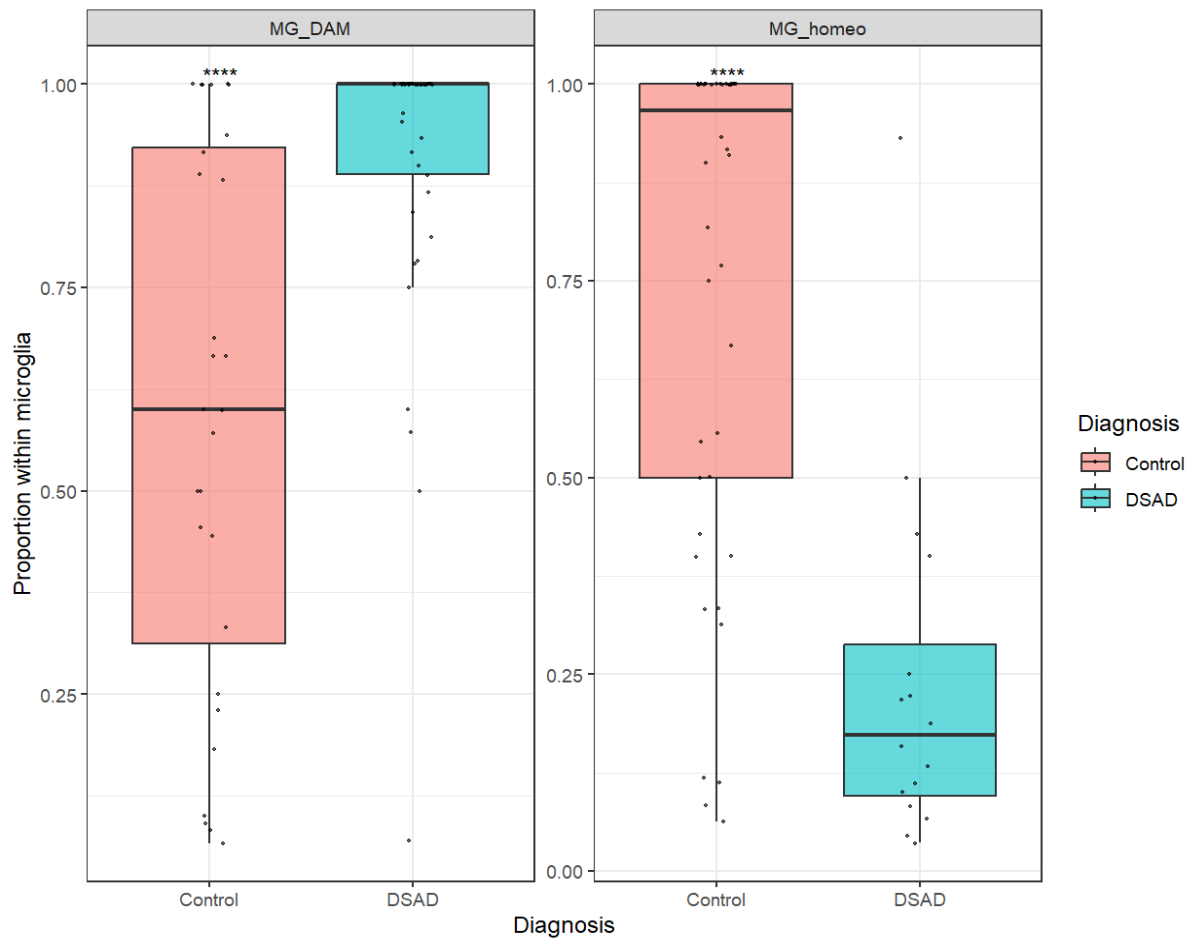
每个MG_state在Control vs DSAD中的丰度差异:以样本为单位比较,而不是直接数细胞
mg_state_comp <- mg_pure@meta.data %>%
group_by(SampleID, Diagnosis, MG_state2) %>%
summarise(n = n(), .groups = "drop") %>%
group_by(SampleID) %>%
mutate(freq = n / sum(n))
ggplot(mg_state_comp, aes(x = Diagnosis, y = freq, fill = Diagnosis)) +
geom_boxplot(outlier.shape = NA, alpha = 0.6) +
geom_jitter(width = 0.1, size = 0.5, alpha = 0.6) +
facet_wrap(~ MG_state2, scales = "free_y") +
ylab("Proportion within microglia") +
theme_bw() +
stat_compare_means(
method = "wilcox.test",
label = "p.signif"
)
某个“显著state”里面发生了什么/比较不同MG_state之间的marker和富集情况:
- 在AD样本中比例显著升高的MG_DAM这个状态内部,DSAD相比Control还多了哪些额外的表达变化,这些变化都富集了哪个通路
Idents(mg_pure) <- "MG_state2" mg_dam <- subset(mg_pure, idents = "MG_DAM") Idents(mg_dam) <- mg_dam$Diagnosis mg_dam_DE <- FindMarkers( mg_dam, ident.1 = "DSAD", ident.2 = "Control", test.use = "MAST", logfc.threshold = 0.1, min.pct = 0.1, latent.vars = c("nCount_RNA", "Batch", "PMI", "Sex") ) # 由于差异基因很少,就不做后续分析了 - 在MG内比较MG_DAM和MG_homeo的差异基因,看MG_DAM这种状态在路径/功能上到底和MG_homeo有什么不同
mg_state_DE <- FindMarkers( mg_pure, ident.1 = "MG_DAM", ident.2 = "MG_homeo", test.use = "MAST", logfc.threshold = 0.1, min.pct = 0.1 ) mg_state_DE_up <- mg_state_DE %>% dplyr::filter(avg_log2FC > 0, p_val_adj < 0.1) %>% arrange(desc(p_val_adj)) ego <- enrichGO( gene = rownames(mg_state_DE_up), OrgDb = org.Hs.eg.db, keyType = "SYMBOL", ont = "BP", pAdjustMethod = "BH", pvalueCutoff = 0.05, qvalueCutoff = 0.2, minGSSize = 5 ) dotplot(ego, showCategory = 20) + theme_bw()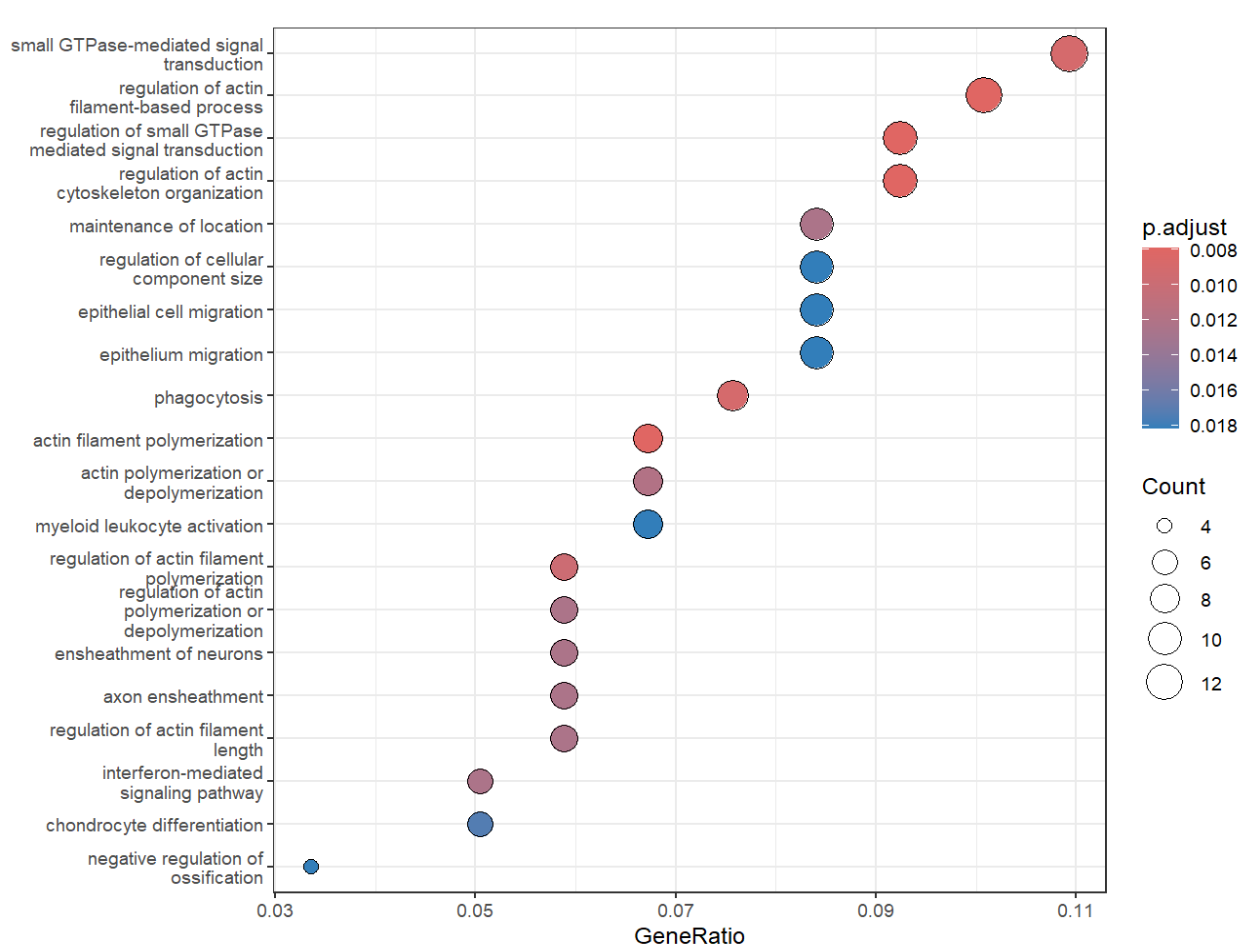
补充:原文中使用的MiloR——基于KNN neighbors,不依赖cluster边界,而是直接在连续的表达空间上做差异丰度测试,相比上面的简化版流程,对细胞分类划分的更细,更容易发现“哪个局部区段变化最大”(如果变化只发生在某个细胞类型/state里的一小部分,但cluster划分时把这一大部分都当成一个整体,就没法看出这一小部分细胞的变化,而Milo能避免过度依赖人工划的cluster边界)
- 当样本量较大,或者单个细胞类型里有明显的连续轨迹(而不是几个清晰cluster)、变化可能发生在过度细胞(intermediate state)中,或者想严格考虑批次效应这些协变量的时候,就应用MiloR
补充:作者的注释——其实作者已经在sn@meta.data对各类细胞进行注释了,不仅是前面使用过的”cell_type”,还有更细的”annotation”/”subtype”/”cell_identity”等,因此在找MG的state步骤中,实际上其实是不需要我们自己手动分类标明的
与hERV
先找到GSE233208的说明
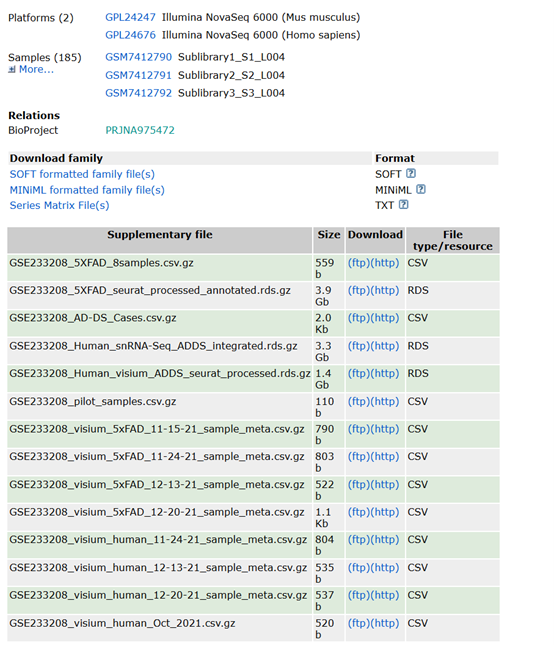
- 最上面的SOFT/MINiML:这个series(GSE233208)的说明、每个平台(GPL…)、每个样本(GSM…)的元数据,有些数据类型还会带表达矩阵(更多见于微阵列、bulk RNA-seq)。一般用于GEO2R等工具,或者想批量看元数据时解析用,对于目前的snRNA-seq项目来说用处不大
- 最上面的Series Matrix:通常是把表达矩阵和样本注释打包成一个大表,多见于微阵列/bulk RNA-seq,但这个项目中作者已把单细胞/空间转录组数据整理成RDS/CSV形式放在Supplementary file
-
GSE233208_5XFAD_8samples.csv.gz:5XFAD小鼠那一部分的样本信息表(8个样本) -
GSE233208_5XFAD_seurat_processed_annotated.rds.gz:小鼠5xFAD脑(AD模型)的数据,作者已经在R里做完预处理、聚类、注释,存成一个Seurat对象 -
GSE233208_AD-DS_Cases.csv.gz:人类DSAD/对照患者的病例信息表(年龄、性别、诊断等),后面做差异分析、分组(Control vs DSAD等)要用的meta表 -
GSE233208_Human_snRNA-Seq_ADDS_integrated.rds.gz:人类单核RNA-seq整合Seurat对象,包括29个前额叶皮质样本(DSAD/对照)的snRNA-seq,已经做完质控、归一化、整合、聚类、UMAP、细胞类型注释等 -
GSE233208_Human_visium_ADDS_seurat_processed.rds.gz:人类Visium空间转录组的Seurat对象,同样已经处理成Seurat对象,用于空间图谱、和snRNA整合等分析 -
GSE233208_pilot_samples.csv.gz:一些试验性pilot样本的meta表,规模很小,一般可以先忽略 -
GSE233208_visium_5xFAD_*_sample_meta.csv.gz:5XFAD小鼠Visium空间转录组的样本信息表(不同测序批次/日期) -
GSE233208_visium_human_*_sample_meta.csv.gz:人类Visium空间转录组的样本信息表 - 注意页面最下面的”Raw data are available in SRA”、”Processed data are available on Series record”,就是说原始读段在SRA上下载,处理好后的Seurat/RDS/CSV就在这个GEO Series页面
点击页面最下方的”SRA Run Selector”,进入PRJNA975472的SRA Run Selector界面,Assay Type选rna-seq,之后发现Organism只剩下人类,tissue和source_name只有prefrontal cortex,这时剩下的41条数据理论上就是我们想找的数据
- 其实是没错的,因为根据文章和GEO说明,这个项目的snRNA-seq部分,就是对DSAD/对照患者的前额叶皮质(prefrontal cortex)做的单核RNA-seq,差异是在Diagnosis(AD/正常)、年龄、性别等临床信息以及不同细胞类型之间的表达差异上
- diagnosis列全是NA:临床信息在
GSE233208_AD-DS_Cases.csv中,不在SRA run中显示;而且这套snRNA-seq的每个run(SRR)都包括多个样本,这多个样本的diagnosis状况可能不同,所以只能存在那个csv表中
选200G~300G的数据,不过这些数据理应包含足够的DSAD/正常样本,这个情况就只能跑完全套流程后再到Seurat对象中检查
snRNA-seq和普通的RNA-seq的区别:snRNA-seq多了两个关键维度——cell barcode(CB)标识每条read属于哪个细胞,UMI(UB)标识每条reads来自哪个原始转录本分子,因此不能直接套用前面的bowtie2+Telescope。大概有两种思路
- 找一个普通的基因gtf,再找一个针对hERV的gtf,合并,直接用STARsolo+cellranger或Parse Biosciences官方pipeline来计数,得到
matrix.mtx/features.tsv/barcodes.tsv这三个文件,然后构建Seurat对象,后面分析时可以把这个大的Seurat对象拆成“基因(RNA)”和“hERV”两个assay,分别进行分析 - 先用常规方法计数普通基因,再用STARsolo+Stellarscope(位点级)/scTE(家族级)计数hERV,最终得到两个计数矩阵和注释信息(需确保这两个矩阵的列名——barcode完全一致,行名就是各自的features——基因/hERV位点),用基因计数矩阵构建Seurat对象,然后再把hERV计数矩阵作为一个新的assay加上去
主要目标:在某个细胞类型内,比较DSAD vs Control的hERV激活
- 只用hERV?hERV+基因?先都做了再说
- 12月完成fastq->计数矩阵,1~2月Seurat分析
关于如何在细胞注释/细胞轨迹中观测hERV的重调变化:先用“正常基因”做好细胞注释和轨迹,再把hERV当成一个额外“分子特征”,往这些注释和轨迹上去“上色”和“切分”(?)