使用我的GSE157827数据进行初步分析-4
other-other2026.3.11-2026.3.18研究进展
AD数据汇总
AD的scRNA/snRNA-seq:
- GSE138852:我第一次用的测试数据,snRNA-seq,内嗅皮层(Entorhinal Cortex),共78.18G,碱基数153.78G,样本量8个,使用10X Genomics,来自《A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation》
- GSE157827:我正在使用的数据,snRNA-seq,前额皮层(prefrontal cortex),共593.81G,碱基数2.02T,样本量21个,使用10X Genomics,来自《Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease》,每个样本的具体信息(组别、性别、年龄、APOE等)
- GSE233208:原定在毕设中使用的数据,包括小鼠5xFAD脑和人类的snRNA-seq和空间转录组数据,并有作者整合好的Seurat对象;其中人类snRNA-seq数据共732.66G,碱基数2.29T,样本数96个,前额皮层(prefrontal cortex)+后扣带皮层(posterior cingulate cortex),但使用一个未开源的测序+计数pipeline(splitpipe),很难自行从原始的fastq进行gene和hERV的计数(详见我的研究记录中
GSE233208部分)。来自《Spatial and single-nucleus transcriptomic analysis of genetic and sporadic forms of Alzheimer’s disease》,每个样本的具体信息 - synapse举办的一个比赛SEA-AD DREAM Challenge - Predicting Alzheimers Pathology from snRNA-seq Data:总共100多G的超大snRNA-seq数据集,但应该是计数完的表达矩阵,似乎没看到原始fastq文件
- GSE174367:snATAC-seq/snRNA-seq以及bulk RNA-seq,其中snRNA-seq共19个样本,bulk RNA-seq近200个样本,10xGenomics Chromium V3测序,前额皮层(prefrontal cortex),来自《Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease》
- syn52293417:snATAC-seq/snRNA-seq,10x Genomics,数据库中总共427个样本(文章中似乎用了92个),前额皮层(prefrontal cortex),来自《Epigenomic dissection of Alzheimer’s disease pinpoints causal variants and reveals epigenome erosion》
- GSE289721:针对小胶质细胞的scRNA-seq,研究how AD GWAS hits influence human microglial inflammatory responses,来自《High throughput identification of genetic regulators of microglial inflammatory processes in Alzheimer’s disease》
其它暂时应该用不上的数据:
- GSE207099:CROPSeq of Putative AD- and PSP-associated cis-Regulatory Regions in iPSC-derived Neurons and Microglia,来自《Functional regulatory variants implicate distinct transcriptional networks in dementia》
- 《Single-cell genomics and regulatory networks for 388 human brains》:用了很多个数据集,详细信息 | 对应下载链接
- EPI数据(enhancer-promoter interaction):
- eQTL数据(定量性状基因位点):
- 张江脑库两个AD队列与telescope的herv的gtf比较,得到与herv有overlap的结构变异,可以筛选在AD/NC中显著多发生结构变异的herv:下载
用管家基因来验证测序深度对表达量计数的影响
hk_genes <- c("ACTB", "GAPDH", "RPL13A", "RPLP0", "EEF1A1", "PPIA")
hk_genes <- intersect(hk_genes, rownames(seu))
meta <- seu@meta.data %>%
rownames_to_column("cell") %>%
select(cell, group, nCount_RNA) %>%
filter(group %in% c("NC", "AD")) %>%
mutate(
group = factor(group, levels = c("NC", "AD")),
log10_nCount_RNA = log10(nCount_RNA + 1)
)
cells_use <- meta$cell
expr_mat <- GetAssayData(seu, assay = "RNA", slot = "data")[hk_genes, cells_use, drop = FALSE]
df_long <- lapply(seq_along(hk_genes), function(i) {
data.frame(
cell = cells_use,
gene = hk_genes[i],
expr = as.numeric(expr_mat[i, ]),
stringsAsFactors = FALSE
)
}) %>%
bind_rows() %>%
left_join(meta, by = "cell")
stat_res <- df_long %>%
group_by(gene) %>%
summarise(
NC_median = median(expr[group == "NC"], na.rm = TRUE),
AD_median = median(expr[group == "AD"], na.rm = TRUE),
diff_median = AD_median - NC_median,
p_wilcox = wilcox.test(expr ~ group)$p.value,
rho_nCount = suppressWarnings(cor(expr, log10_nCount_RNA,
method = "spearman",
use = "complete.obs")),
.groups = "drop"
) %>%
mutate(
p_wilcox_adj = p.adjust(p_wilcox, method = "BH"),
celltype = "All"
) %>%
arrange(p_wilcox_adj)
stat_by_celltype <- df_long %>%
group_by(celltype, gene) %>%
summarise(
NC_median = median(expr[group == "NC"], na.rm = TRUE),
AD_median = median(expr[group == "AD"], na.rm = TRUE),
diff_median = AD_median - NC_median,
p_wilcox = tryCatch(wilcox.test(expr ~ group)$p.value, error = function(e) NA_real_),
rho_nCount = suppressWarnings(cor(expr, log10_nCount_RNA,
method = "spearman",
use = "complete.obs")),
.groups = "drop"
) %>%
group_by(celltype) %>%
mutate(p_wilcox_adj = p.adjust(p_wilcox, method = "BH")) %>%
ungroup()
write.csv(bind_rows(stat_res, stat_by_celltype), file = "C:\\Users\\17185\\Desktop\\hERV_calc\\GSE157827\\my_res\\ncount_gene.csv", row.names = F)
# AD和NC中这些管家基因的表达分布
ggplot(df_long, aes(x = group, y = expr, fill = group)) +
geom_violin(scale = "width", trim = TRUE) +
geom_boxplot(width = 0.12, outlier.size = 0.2, fill = "white") +
facet_wrap(~ gene, scales = "free_y", ncol = 3) +
stat_compare_means(method = "wilcox.test", label = "p.format") +
theme_classic(base_size = 12)
# 这些管家基因表达和nCount_RNA是否仍明显相关
ggplot(df_long, aes(x = log10_nCount_RNA, y = expr)) +
geom_point(size = 0.2, alpha = 0.25) +
geom_smooth(method = "lm", se = FALSE) +
facet_wrap(~ gene, scales = "free_y", ncol = 3) +
theme_classic(base_size = 12)
标准化后,测序深度的影响被减弱了:
- 虽然选的是比较常见的基因,但大部分中位数还是0(有超过一半的细胞没有表达该基因),因此diff_median就不太能作为评估标准
- 相关性系数rho_nCount几乎都在0.3以下,但测序深度的影响没有被完全消除
- 似乎没找到什么有效的方法来证明测序深度的影响是否存在,毕竟使用的流程是Seurat官方推荐的,只能说大家都是这么做的,而且常见的都是对普通基因这种计数比较稳定的,有点影响很容易就被后面DE分析时回归掉,像hERV这种稀疏计数并不常见
如何解决:
- 在原论文中,作者确实用了
IntegrateData来整合所有样本,然后再做PCA、UMAP和聚类。但要注意,去批次效应主要影响降维/聚类(细胞类型注释),而不是差异表达分析(DE分析使用的是标准化后的表达量,而不是去批次效应/整合后的数据) ScaleData在预处理阶段直接改写表达矩阵,不能把orig.ident(样本因素)直接作为参数vars.to.regress的值,相当于在做PCA/UMAP/聚类前,就尝试把donor差异从表达里整体减掉,而这组数据里样本和疾病状态直接相关,有风险把真正的疾病信号也一起削掉- “在后续差异表达分析时,把nCount_RNA作为协变量回归”也算是正常方法,虽然更广泛更科学的方法是pseudobulk(以样本而不是细胞为单位进行分析)
- 在
FindMarkers时用latent.vars=c("nCount_RNA","percent_mito")(而不是之前的latent.vars=c("orig.ident","nCount_RNA","percent_mito"),原因同上,防止把真正的疾病效应消除。如果是同一个donor但有两种不同疾病状态,就可以加上)
- 在
- “细胞亚群聚类时,某一个cluster中会有某一个或者几个样本占很大部分”有可能是正常现象
- 如果某个亚群本来就是一种比较少见、容易受疾病或个体状态影响的细胞状态,那么它在少数donor里明显富集是完全可能的,尤其在样本数本来就不多的情况下
- 但仍需根据
nCount_RNA/percent_mito来判断是不是异常细胞聚集 SCTransform+integration的处理不一定能完全减少这种现象,比如上面说的“某亚群真的是一种特殊状态”,去批次效应就不一定把它抹平,有时它也不该被抹平;过度整合有可能把真实生物差异也削弱
因此最后还是觉得从头走一遍去批次效应的流程比较好,除了和论文对齐、尽量消除测序深度的影响外,更主要还是觉得数据预处理的流程(尤其是hERV表达量计算的步骤)更改了好几次,可能改的有点乱,为了最后汇总代码也应该从头运行一遍
- 按样本拆分后,每个样本分别做
SCTransform,之后再IntegrateData:如果直接SCTransform,就相当于把所有细胞当成一个整体,用一个模型统一做归一化/方差稳定化,适用于单一样本/样本很少且批次效应不明显/不准备做integration的情况;如果想减少样本来源对聚类的影响,想让同类细胞更多地按细胞类型聚在一起,就还是分别SCTransform- 后续的DE分析使用
"SCT"作为DefaultAssay:"integrated"的assay是“让不同样本更可比”,不是保留原始生物表达幅度
- 后续的DE分析使用
- 后来实际运行了一下
SCTransform+integration流程,可能是因为服务器内存不够,最后还是改成了NormalizeData+harmony流程,得到的结果与之前的差的不是很多,不过确实能看到样本分布更均匀了- 除了
SCTransform+integration流程有亿点点耗时耗内存之外,最主要的原因是我的hERV是用的仿NormalizeData的标准化方法,为了后面hERV~gene共表达分析时更准确合理,普通基因就也用NormalizeData了 - 为什么不把hERV也改成仿SCT的处理方式:SCT主要是为常规基因表达设计和验证的,而hERV矩阵更稀疏、特征数更少,而且计数来源于重复序列重分配结果,不完全等同于普通基因UMI计数,不太清楚是否符合SCT的建模假设。还是先把gene和hERV放在同一背景下比较稳定、比较可实现
- 除了
- 关于MAST方法协变量设置的问题:是否加上协变量
nCount_RNA+percent_mito?按理论来讲,MAST方法内部已经设置了和测序深度相关的指标,所以如果再加上nCount_RNA可能会有过度校正的风险(参考);如果是其它的可以设置协变量的方法,其实是可以加的 - 选某一细胞类型再做亚群聚类:
- 可以继续用
SCTransform+integration,就DefaultAssay(astro) <- "integrated",然后RunPCA等,这种方法最简单,但如果全局整合时细胞类型内部细粒度差异没有被很好保留,亚群分辨率可能一般 - 最普通的方法(第一次我采用的方式):
NormalizeData + FindVariableFeatures + ScaleData + PCA + UMAP + 聚类,适用于这个细胞类型内部的样本效应不算特别严重的情况 - 还可以沿用之前的思路
NormalizeData+harmony(现在我采用的方式) - 最严格的方法:先取出目标细胞类型,再按样本拆分,只在这个细胞类型内部做
SplitObject + SCTransform + IntegrateData,有时会比第一种方法更能保留细胞类型内部细粒度结构,适用于这个细胞类型内部仍明显按样本分开的情况
- 可以继续用
gtf文件-hERV家族名
所有位点都有gene行(都有intModel),而所有的intModel都是以-int结尾的,同时大多数位点exon行的geneRegion都分为internal和ltr(前病毒结构分为两端LTR与内部区internal,env/gag/pol都包含在这个内部区里),internal和ltr对应的repName可能不同,但这不体现在位点名上(所有的位点名都是”ERV316A3_1p36.33”这种,不标识具体是int还是ltr)。因此在本研究的位点/亚家族聚合分析中,-int后缀不提供额外的分层信息
- 去掉
-int后缀:去掉的不是“生物学上的internal信息”本身,而只是把它从亚家族命名里去掉,让亚家族标签更简洁、更适合聚合统计。LTR/internal的结构差异并没有消失,因为它仍然可以在exon的geneRegion里看到 - “不体现在位点名上”更准确的说法:位点名本身不编码LTR/internal信息,因此
-int在当前gene-level位点聚合中主要体现的是注释来源(internal model),而不是可进一步区分的位点层级类别
继续之前的细胞类型-hERV家族水平重调分析
感觉还得是用普通基因来分亚群,如果用hERV某家族的表达量来分,感觉受nCount_RNA影响太大了,就像之前的那样,分完之后发现hERV表达量高的都是nCount_RNA高的
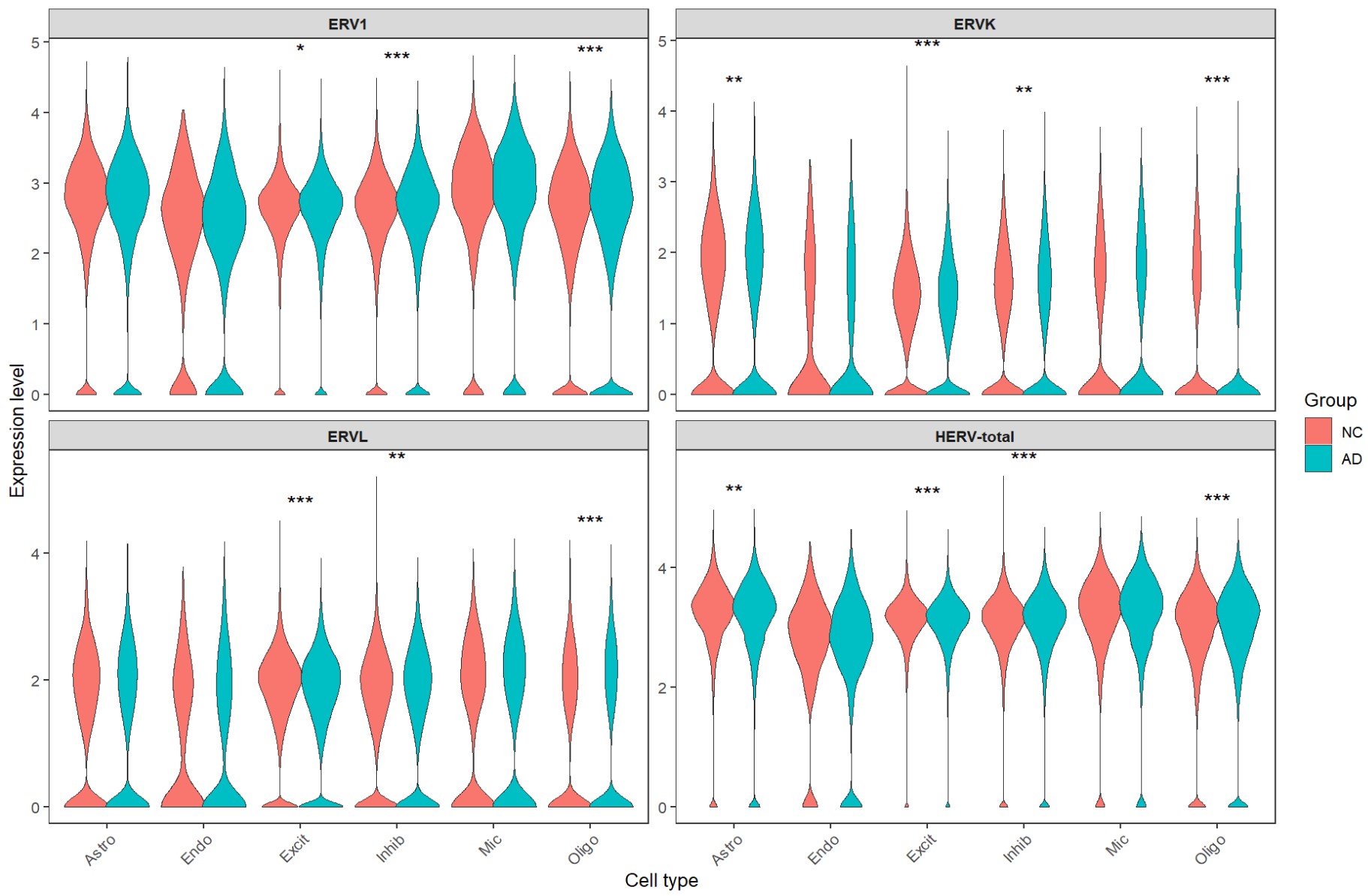
调整协变量后的新差异表达结果
总的来说,其实相差不是很大,而且变化的主要是p值
全体细胞的超家族:
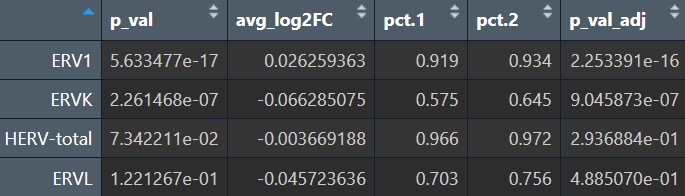
各细胞类型的超家族:
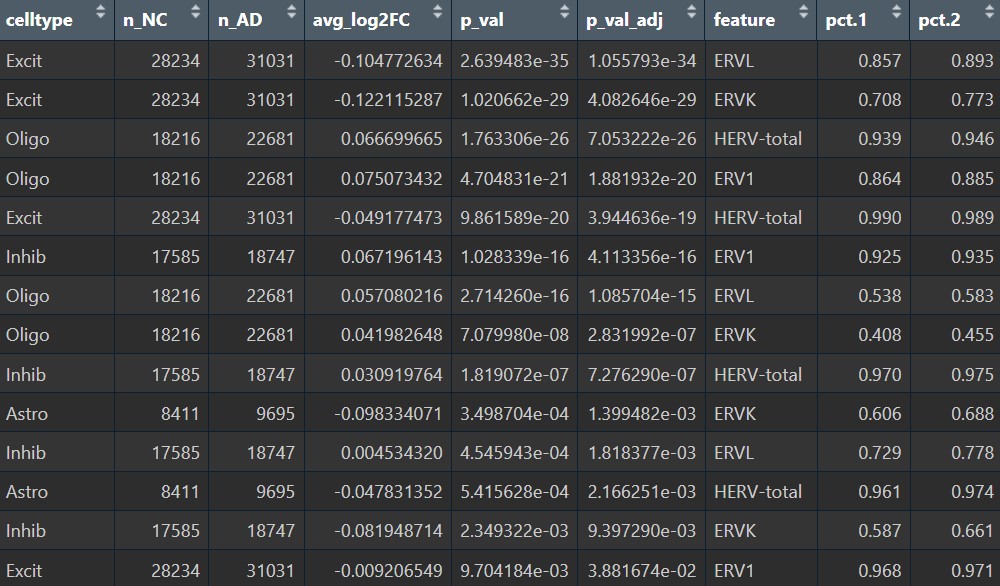
inhib类型在这步里变得比较显著,也许也可以作为分析对象之一
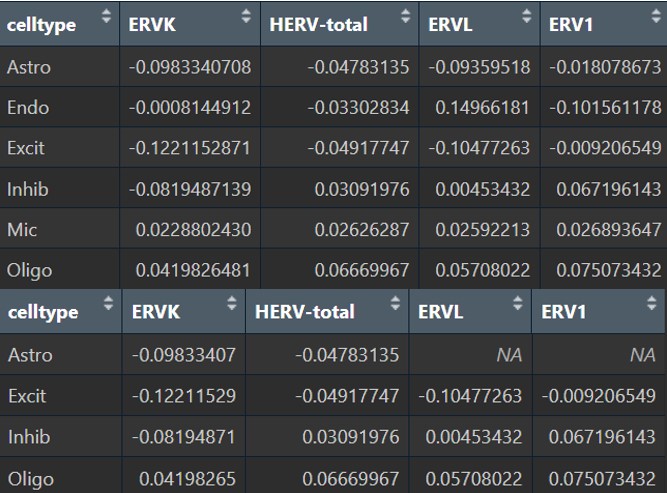
各细胞类型的亚家族:
这里的变化就稍微有点大了,比如astro的HERVK的p值就变成了1(而原来是<0.05的),然后HERV3这个家族看起来表达量很高的样子
调整excit的分辨率再看看表达量热图
除了改分辨率之外,对于目标hERV家族的选取也尽可能宽松一些,防止有些hERV家族只在某些亚群中特异表达而被过滤掉
excit$excit_subcluster <- excit$RNA_snn_res.0.1
# excit$excit_subcluster <- excit$RNA_snn_res.0.3
Idents(excit) <- excit$excit_subcluster
DimPlot(excit, reduction = "umap", group.by = "excit_subcluster", label = TRUE)
res_subfamily <- read.csv("C:\\Users\\17185\\Desktop\\hERV_calc\\GSE157827\\my_res\\hERV_family.csv")
res_excit_focus <- res_subfamily %>%
filter(
celltype == "excit",
pmax(pct.1, pct.2) >= 0.10, # 至少一组有一定检出
pmin(pct.1, pct.2) >= 0.02 # 另一组不要过于稀疏
) %>%
arrange(desc(abs(avg_log2FC)), p_val_adj)
down_subfam <- res_excit_focus %>% # 下调
filter(avg_log2FC < 0) %>%
arrange(avg_log2FC) %>%
pull(subfamily)
up_subfam <- res_excit_focus %>% # 上调
filter(avg_log2FC > 0) %>%
arrange(desc(avg_log2FC)) %>%
pull(subfamily)
# 画图代码不变


感觉不如之前的那版突出,但之前的那版存在一个小问题,就是cluster3(HERV3高表达)的样本集中在两个样本内,可能会有些不稳定?
Oligo
oligo <- subset(seu, subset = celltype == "Oligo")
DefaultAssay(oligo) <- "RNA"
oligo <- FindVariableFeatures(
oligo,
selection.method = "vst",
nfeatures = 2000
)
oligo <- ScaleData(
oligo,
features = VariableFeatures(oligo),
vars.to.regress = c("nFeature_RNA", "percent_mito")
)
oligo <- RunPCA(
oligo,
features = VariableFeatures(oligo),
)
ElbowPlot(oligo, ndims = 50)

pc.num <- 1:20
oligo <- RunUMAP(
oligo,
dims = pc.num
)
DimPlot(oligo, group.by = "group") | DimPlot(oligo, split.by = "group")

oligo的和excit的不同,AD/NC分布还是挺大的
oligo <- FindNeighbors(
oligo,
dims = pc.num
)
oligo <- FindClusters(
oligo,
resolution = c(0.01,0.05,0.1,0.2,0.3,0.4,0.5)
)
clustree(oligo@meta.data, prefix = "RNA_snn_res.")

oligo$oligo_subcluster <- oligo$RNA_snn_res.0.3
Idents(oligo) <- oligo$oligo_subcluster
DimPlot(oligo, reduction = "umap", group.by = "oligo_subcluster", label = TRUE)
saveRDS(
oligo,
file = "C:\\Users\\17185\\Desktop\\hERV_calc\\GSE157827\\my_data\\GSE157827_oligo.rds",
compress = "xz"
)

res_oligo_focus <- res_subfamily %>%
filter(
celltype == "Oligo",
!is.na(p_val_adj),
p_val_adj < 0.05,
abs(avg_log2FC) >= 0.05,
pmax(pct.1, pct.2) >= 0.10,
pmin(pct.1, pct.2) >= 0.02
) %>%
arrange(desc(abs(avg_log2FC)), p_val_adj)
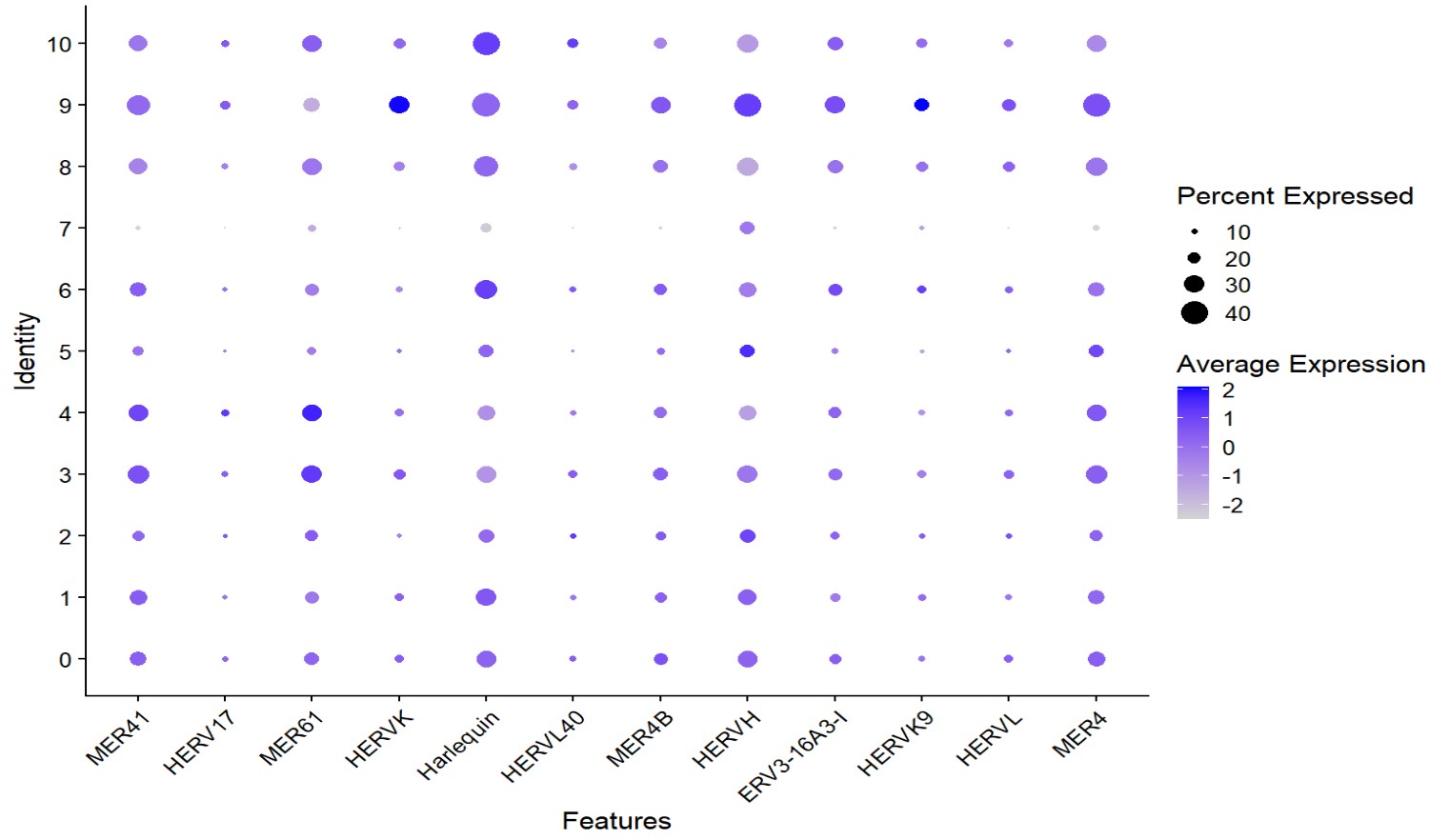
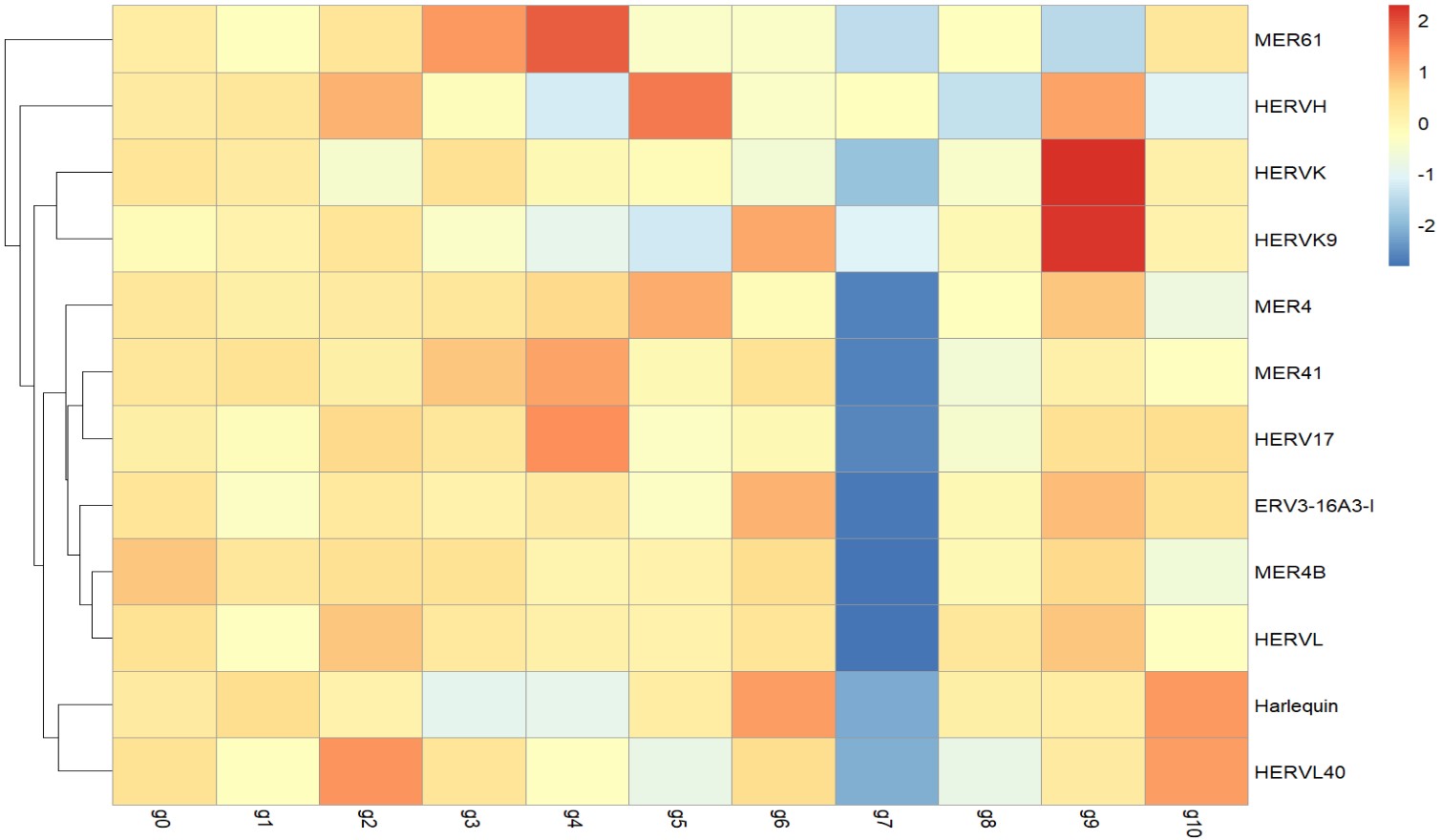
似乎cluster 9的HERVK表达较高
> table(oligo$orig.ident, oligo$oligo_subcluster)[, "9"]
AD1 AD10 AD13 AD19 AD2 AD20 AD21 AD4 AD5 AD6 AD8 AD9 NC11
127 17 1 5 205 113 5 82 25 0 37 22 0
NC12 NC14 NC15 NC16 NC17 NC18 NC3 NC7
107 22 2 10 15 119 23 22
> table(oligo$group, oligo$oligo_subcluster)[, "9"]
NC AD
320 639
样本分布比较均匀,并且AD细胞较多,感觉可以作为下一步的主要研究对象
Astro

pc.num <- 1:20
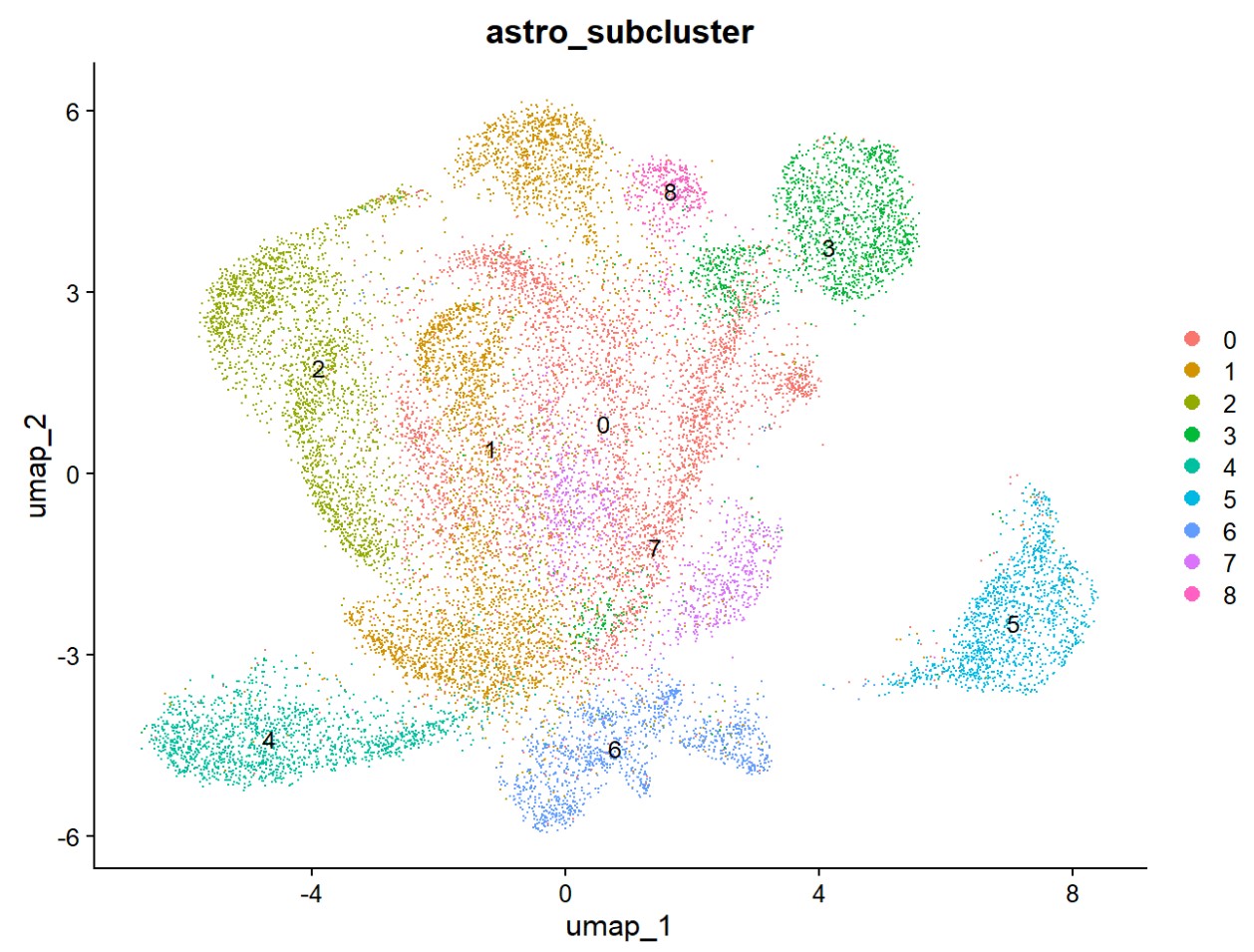
astro$astro_subcluster <- astro$RNA_snn_res.0.2
res_astro_focus <- res_subfamily %>%
filter(
celltype == "Astro",
!is.na(p_val_adj),
abs(avg_log2FC) >= 0.05,
pmax(pct.1, pct.2) >= 0.10,
pmin(pct.1, pct.2) >= 0.02
) %>%
filter(
!subfamily %in% c("HUERS-P1", "Harlequin", "PRIMA4"),
!grepl("^(MER|ERV)", subfamily)
) %>%
arrange(desc(abs(avg_log2FC)), p_val_adj)
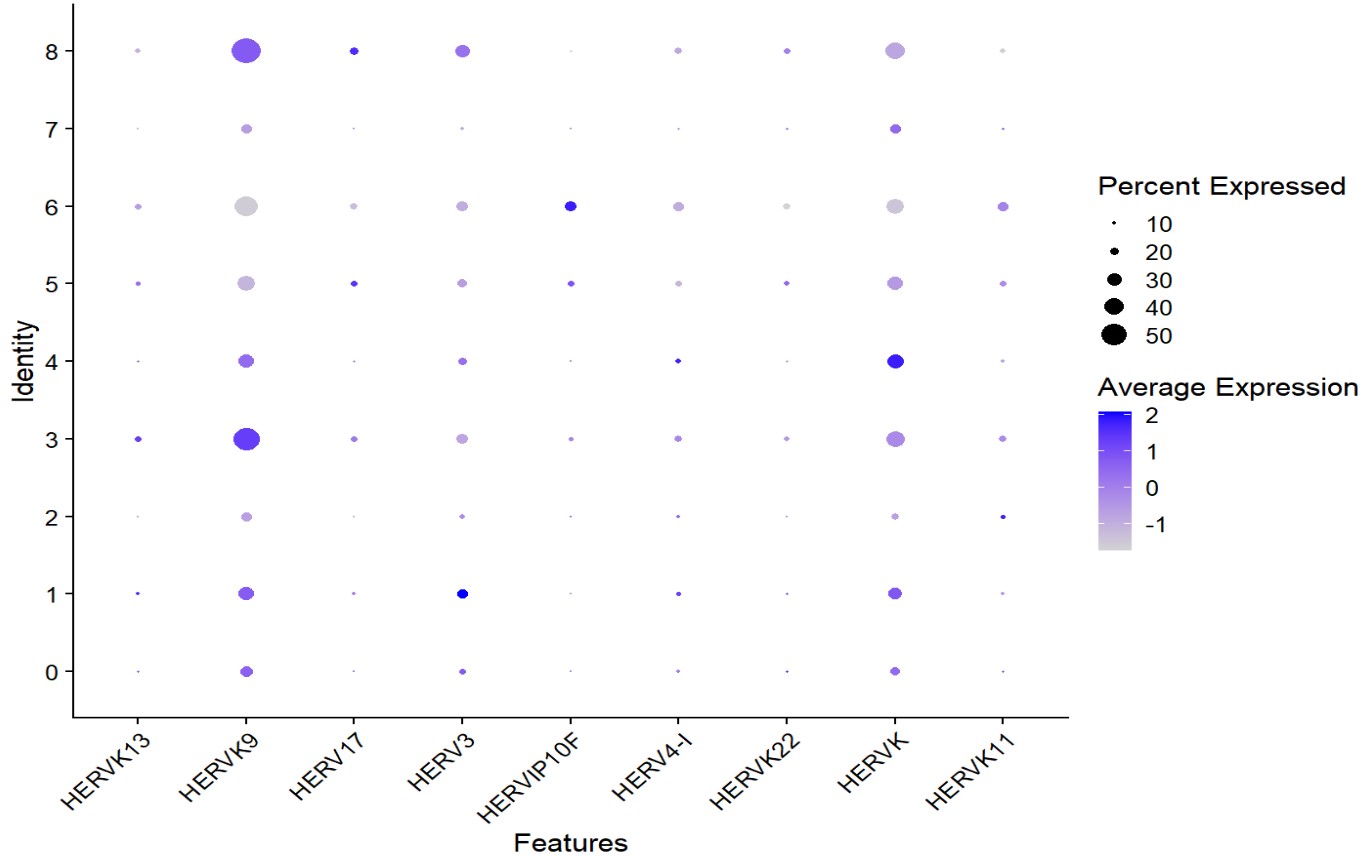
似乎cluster 4的HERVK表达较高,2的HERVK11表达较高,1的HERV3表达较高
> table(astro$orig.ident, astro$astro_subcluster)[, "4"]
AD1 AD10 AD13 AD19 AD2 AD20 AD21 AD4 AD5 AD6 AD8 AD9 NC11 NC12 NC14 NC15 NC16 NC17 NC18 NC3 NC7
8 17 0 0 1497 7 9 1 6 1 2 11 0 0 1 0 1 13 1 10 0
> table(astro$group, astro$astro_subcluster)[, "4"]
NC AD
26 1559
> table(astro$orig.ident, astro$astro_subcluster)[, "2"]
AD1 AD10 AD13 AD19 AD2 AD20 AD21 AD4 AD5 AD6 AD8 AD9 NC11 NC12 NC14 NC15 NC16 NC17 NC18 NC3 NC7
0 2 0 16 4 1289 1084 8 0 0 7 5 0 1 3 48 7 28 6 127 0
> table(astro$group, astro$astro_subcluster)[, "2"]
NC AD
220 2415
> table(astro$orig.ident, astro$astro_subcluster)[, "1"]
AD1 AD10 AD13 AD19 AD2 AD20 AD21 AD4 AD5 AD6 AD8 AD9 NC11 NC12 NC14 NC15 NC16 NC17 NC18 NC3 NC7
135 1019 2 13 98 25 73 11 196 0 2 43 1 195 61 49 11 1541 55 45 347
> table(astro$group, astro$astro_subcluster)[, "1"]
NC AD
2305 1617
可以看到4和2两个聚类确实是AD的细胞比较多,但样本还是很集中的,不过如果是因为这几个样本测序深度高而导致HERVK表达较高,那其它几个subfamily也应该都比较高,但从热图上看到它们并不是这样
Inhib
pc.num <- 1:30
inhib$inhib_subcluster <- inhib$RNA_snn_res.0.2
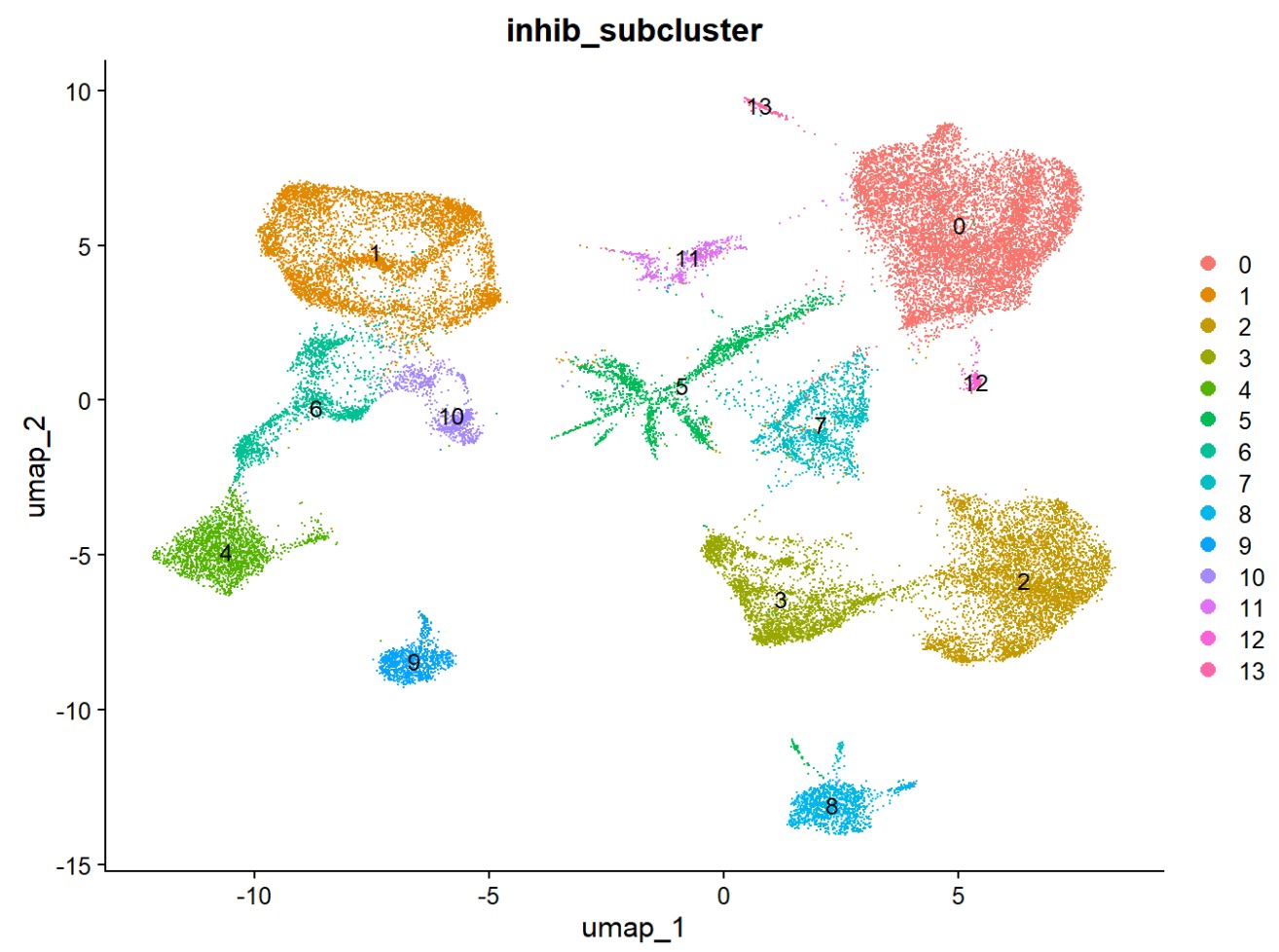
res_inhib_focus <- res_subfamily %>%
filter(
celltype == "Inhib",
!is.na(p_val_adj),
p_val_adj < 0.05,
abs(avg_log2FC) >= 0.05,
pmax(pct.1, pct.2) >= 0.10,
pmin(pct.1, pct.2) >= 0.02
) %>%
arrange(desc(abs(avg_log2FC)), p_val_adj)
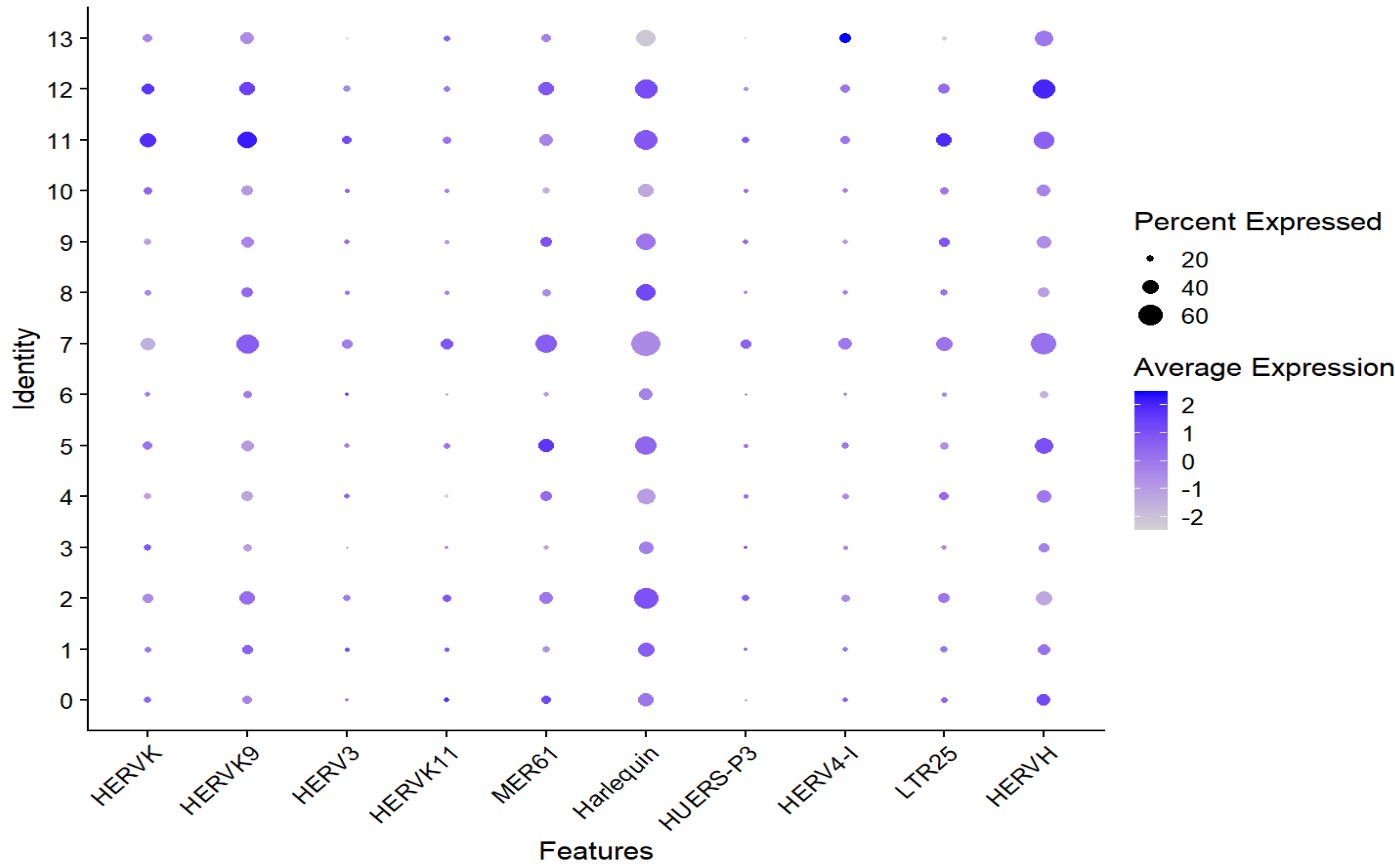
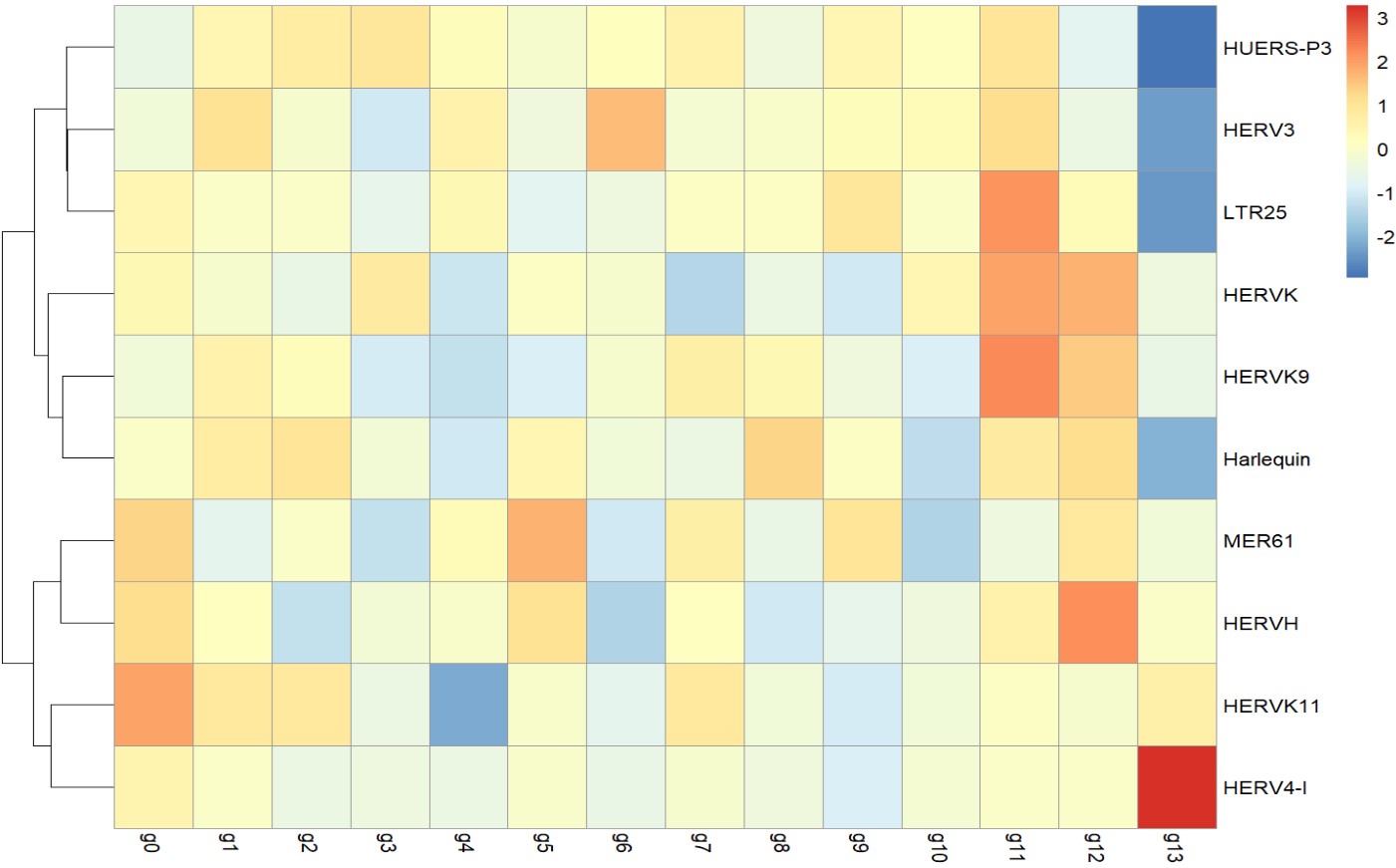
似乎cluster 11的HERVK表达较高
> table(inhib$orig.ident, inhib$inhib_subcluster)[, "11"]
AD1 AD10 AD13 AD19 AD2 AD20 AD21 AD4 AD5 AD6 AD8 AD9 NC11 NC12 NC14 NC15 NC16 NC17 NC18 NC3 NC7
6 50 2 6 64 46 66 35 12 0 1 21 3 36 79 10 16 39 31 17 15
> table(inhib$group, inhib$inhib_subcluster)[, "11"]
NC AD
246 309
这组的结果似乎和oligo的差不多?
亚群内hERV-gene调控网络
在发生herv重调的细胞亚群中,哪些基因/通路受到特异的调控,是否是亚群细胞特异性的,是否与AD相关
以oligo的cluster 3为例:包含1200多个NC和1800多个AD,HERVK表达量较高
- step 1:这个hERV重调亚群本身,哪些基因/通路是这个亚群特异的:即这个亚群和其它亚群对比
- step 2:这个亚群里的基因/通路,哪些又和AD相关
- step 3:看这个亚群内哪些基因与hERVK表达有关(
gene ~ hERV + nCount_RNA + percent_mito)
step 1:
DefaultAssay(oligo) <- "RNA"
Idents(oligo) <- oligo$subcluster
other_ids <- setdiff(levels(Idents(oligo)), "3")
res_rna_3_vs_rest <- FindMarkers(
oligo,
ident.1 = "3",
ident.2 = other_ids,
test.use = "MAST",
# latent.vars = c("nCount_RNA", "percent_mito"),
min.pct = 0.1,
logfc.threshold = 0
) %>%
rownames_to_column("gene") %>%
arrange(p_val_adj, desc(abs(avg_log2FC)))
View(res_rna_3_vs_rest)
sig_3_up <- res_rna_3_vs_rest %>%
filter(!is.na(p_val_adj), p_val_adj < 0.05, avg_log2FC > 0.25) %>%
pull(gene)
sig_3_down <- res_rna_3_vs_rest %>%
filter(!is.na(p_val_adj), p_val_adj < 0.05, avg_log2FC < -0.25) %>%
pull(gene)
write.csv(res_rna_3_vs_rest %>% filter(p_val_adj < 0.05, abs(avg_log2FC) > 0.25), file = "C:\\Users\\17185\\Desktop\\hERV_calc\\GSE157827\\my_res\\oligo_3_test\\res_rna_3_vs_rest.csv")
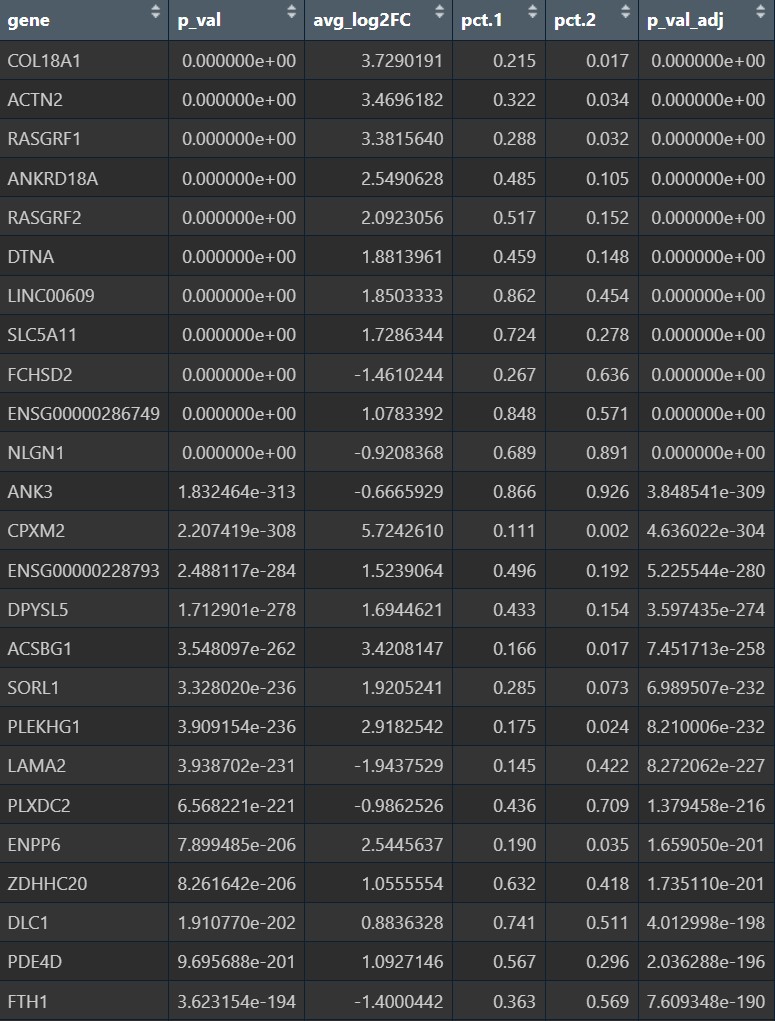
step 2:
oligo3 <- subset(oligo, idents = "3")
Idents(oligo3) <- factor(oligo3$group, levels = c("NC", "AD"))
res_rna_AD_vs_NC_in3 <- FindMarkers(
oligo3,
ident.1 = "AD",
ident.2 = "NC",
test.use = "MAST",
# latent.vars = c("nCount_RNA", "percent_mito"),
min.pct = 0.1,
logfc.threshold = 0
) %>%
rownames_to_column("gene") %>%
arrange(p_val_adj, desc(abs(avg_log2FC)))
View(res_rna_AD_vs_NC_in3)
sig_3_AD_up <- res_rna_AD_vs_NC_in3 %>%
filter(!is.na(p_val_adj), p_val_adj < 0.05, avg_log2FC > 0.25) %>%
pull(gene)
sig_3_AD_down <- res_rna_AD_vs_NC_in3 %>%
filter(!is.na(p_val_adj), p_val_adj < 0.05, avg_log2FC < -0.25) %>%
pull(gene)
write.csv(res_rna_AD_vs_NC_in3 %>% filter(p_val_adj < 0.05, abs(avg_log2FC) > 0.25), file = "C:\\Users\\17185\\Desktop\\hERV_calc\\GSE157827\\my_res\\oligo_3_test\\res_rna_AD_vs_NC_in3.csv")
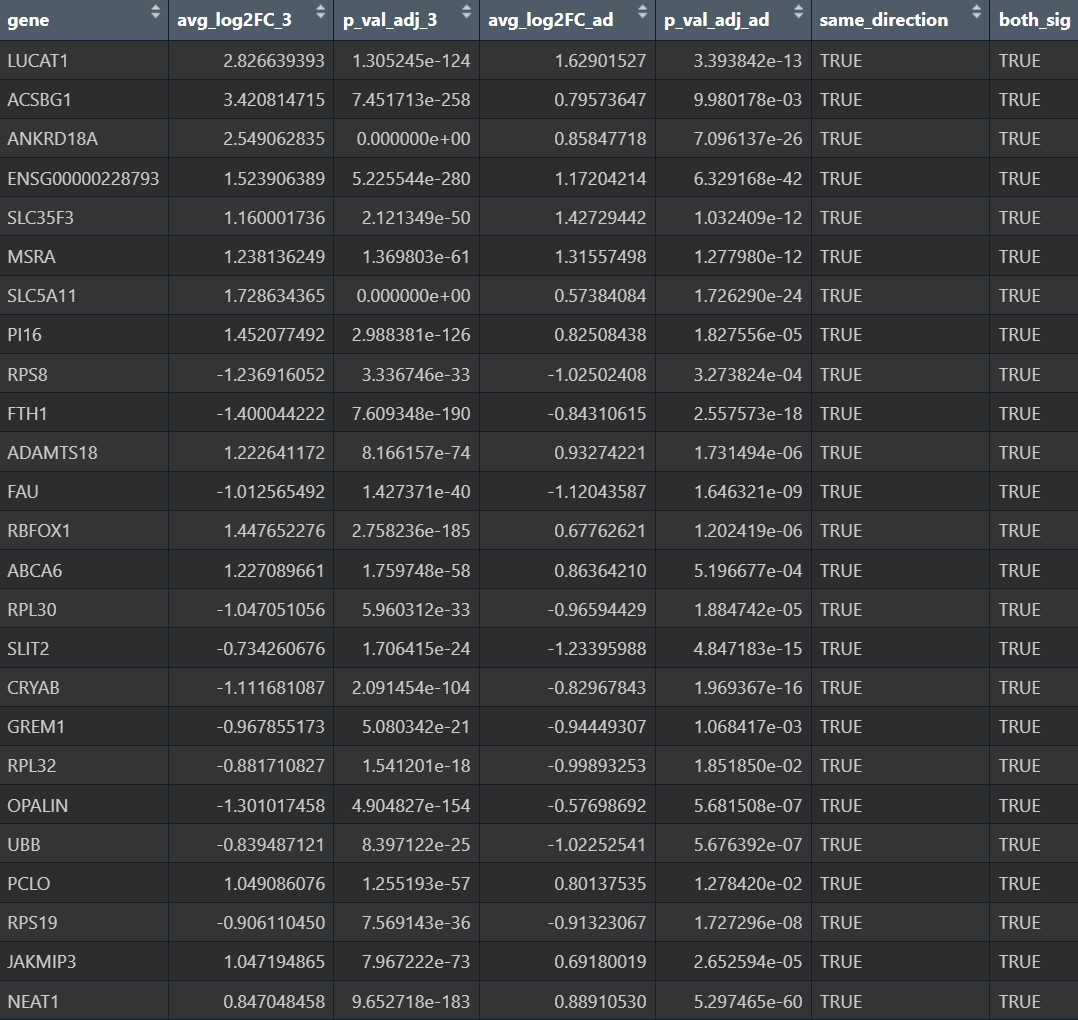
哪些基因既是cluster3特异的,又和AD相关:
genes_3_and_AD_up <- intersect(sig_3_up, sig_3_AD_up)
genes_3_and_AD_down <- intersect(sig_3_down, sig_3_AD_down)
tab_3 <- res_rna_3_vs_rest %>%
dplyr::select(gene, avg_log2FC_3 = avg_log2FC, p_val_adj_3 = p_val_adj)
tab_ad <- res_rna_AD_vs_NC_in3 %>%
dplyr::select(gene, avg_log2FC_ad = avg_log2FC, p_val_adj_ad = p_val_adj)
tab_merge <- inner_join(tab_3, tab_ad, by = "gene") %>%
mutate(
same_direction = sign(avg_log2FC_3) == sign(avg_log2FC_ad),
both_sig = !is.na(p_val_adj_3) & !is.na(p_val_adj_ad) &
p_val_adj_3 < 0.05 & p_val_adj_ad < 0.05
) %>%
arrange(desc(both_sig), desc(same_direction), desc(abs(avg_log2FC_3) + abs(avg_log2FC_ad)))
View(tab_merge)
step 3:
top_overlap <- tab_merge %>%
filter(both_sig, same_direction) %>%
filter(!grepl("^(ENSG|MT-|LINC-)|(AC|AL)[0-9]", gene)) %>%
pull(gene)
DefaultAssay(oligo3) <- "HERV_family"
subfam_use <- c("HERVK", "HERVK3", "HERVK14", "HERVK9", "HERVK13", "HERVK22") # 关注的几个hERV家族
DefaultAssay(oligo3) <- "RNA"
gene_use <- top_overlap
gene_use <- intersect(gene_use, rownames(oligo3[["RNA"]]))
meta3 <- oligo3@meta.data
residualize_vec <- function(y, meta) { # 将测序深度做协变量
fit <- lm(y ~ nCount_RNA + percent_mito, data = meta)
resid(fit)
}
herv_mat <- GetAssayData(oligo3, assay = "HERV_family", slot = "data")[subfam_use, , drop = FALSE]
rna_mat <- GetAssayData(oligo3, assay = "RNA", slot = "data")[gene_use, , drop = FALSE]
herv_resid <- t(apply(as.matrix(herv_mat), 1, residualize_vec, meta = meta3))
rna_resid <- t(apply(as.matrix(rna_mat), 1, residualize_vec, meta = meta3))
res_list <- vector("list", length = nrow(herv_resid) * nrow(rna_resid))
k <- 1
for (i in seq_len(nrow(herv_resid))) {
for (j in seq_len(nrow(rna_resid))) {
x <- as.numeric(herv_resid[i, ])
y <- as.numeric(rna_resid[j, ])
ok <- is.finite(x) & is.finite(y)
x <- x[ok]
y <- y[ok]
if (length(x) < 10 || sd(x) == 0 || sd(y) == 0) {
res_list[[k]] <- data.frame(
subfamily = rownames(herv_resid)[i],
gene = rownames(rna_resid)[j],
rho = NA_real_,
p_val = NA_real_,
n = length(x)
)
} else {
ct <- suppressWarnings(
cor.test(x, y, method = "spearman", exact = FALSE)
)
res_list[[k]] <- data.frame(
subfamily = rownames(herv_resid)[i],
gene = rownames(rna_resid)[j],
rho = unname(ct$estimate),
p_val = ct$p.value,
n = length(x)
)
}
k <- k + 1
}
}
cor_df <- bind_rows(res_list) %>%
mutate(
p_adj = p.adjust(p_val, method = "BH")
) %>%
arrange(p_adj, p_val, desc(abs(rho)))
write.csv(cor_df, file = "C:\\Users\\17185\\Desktop\\hERV_calc\\GSE157827\\my_res\\oligo_3_test\\herv_gene_cor.csv")
cor_df_filt <- cor_df %>%
filter(!is.na(rho), !is.na(p_val)) %>%
filter(p_val < 0.05)
rho_mat <- cor_df_filt %>%
dplyr::select(subfamily, gene, rho) %>%
tidyr::pivot_wider(names_from = gene, values_from = rho) %>%
as.data.frame()
rownames(rho_mat) <- rho_mat$subfamily
rho_mat$subfamily <- NULL
rho_mat <- as.matrix(rho_mat)
row_score <- rowMeans(rho_mat, na.rm = TRUE)
col_score <- colMeans(rho_mat, na.rm = TRUE)
row_score[is.nan(row_score)] <- -Inf
col_score[is.nan(col_score)] <- -Inf
row_ord <- order(row_score, decreasing = TRUE)
col_ord <- order(col_score, decreasing = TRUE)
rho_mat_ord <- rho_mat[row_ord, col_ord, drop = FALSE]
rho_mat_ord[is.na(rho_mat_ord)] <- 0
p <- pheatmap(
rho_mat_ord,
cluster_rows = FALSE,
cluster_cols = FALSE,
# na_col = "grey90",
fontsize_row = 10,
fontsize_col = 5,
angle_col = 45
)
pdf("C:\\Users\\17185\\Desktop\\hERV_calc\\GSE157827\\my_res\\oligo_3_test\\pheatmap.pdf", width = 15, height = 8)
print(p)
dev.off()
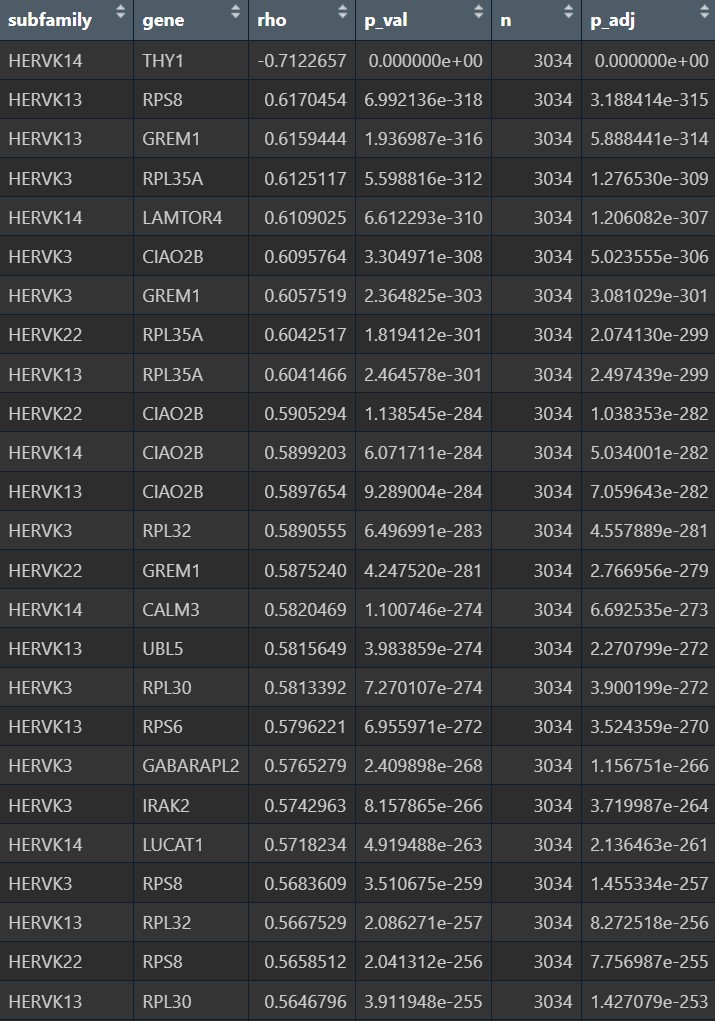
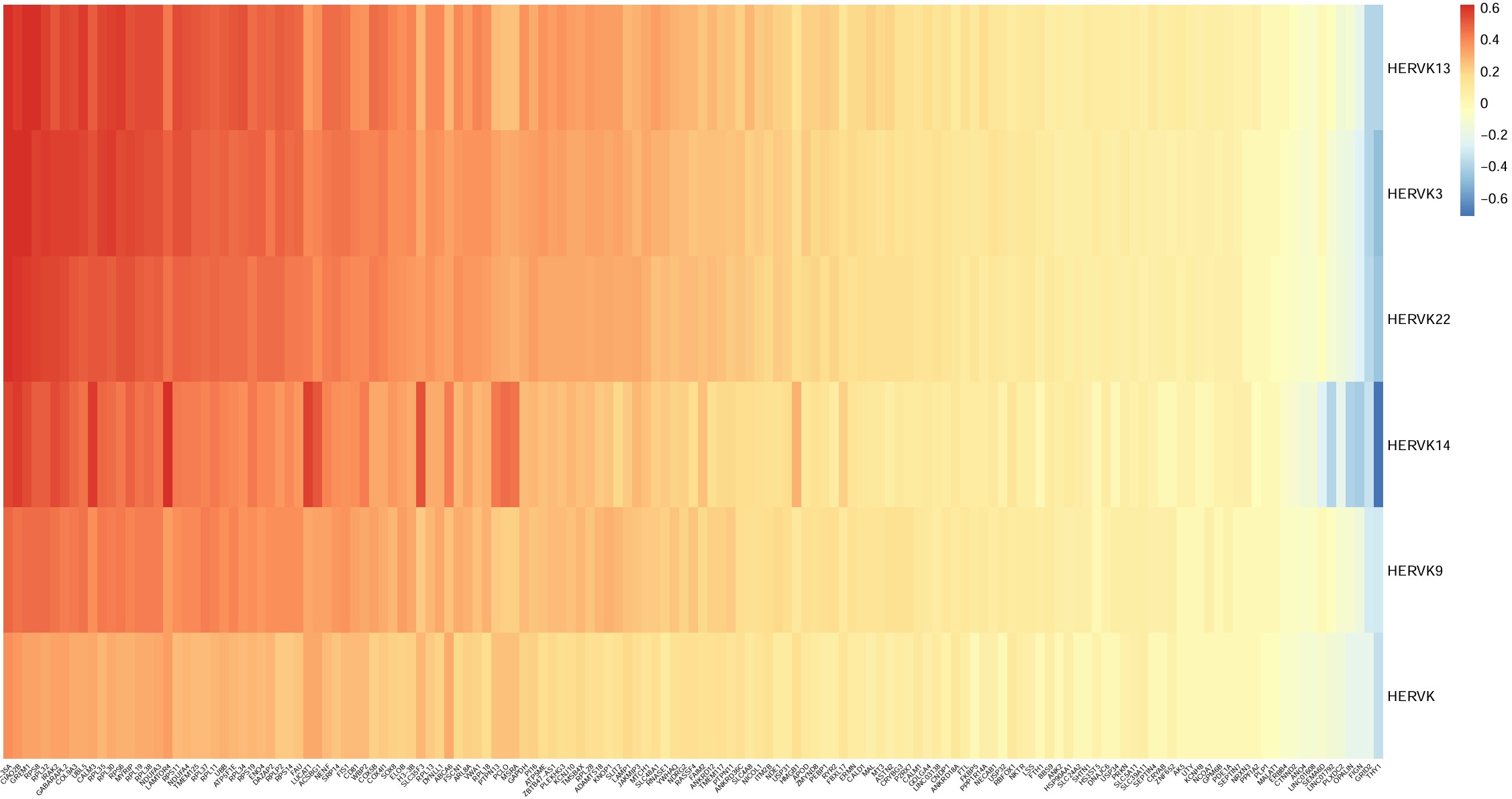
- 正相关:
- 核糖体/蛋白翻译相关:RPL, RPS
- 线粒体呼吸链/能量代谢相关:NDUFA3, NDUFA4, ATP5F1E, COX4I1, COX5B
- 蛋白稳态/自噬-溶酶体/应激相关:GABARAPL2, UBB, LAMTOR4
- 免疫/应激或信号调控相关:IRAK2, CALM3
- 负相关:
- THY1/GRID2:更像神经元相关/成熟状态相关信号
- OPALIN:更偏成熟少突相关标志
下一步研究计划:
- 试一试在细胞大类上做hERV~gene共表达分析:因为之前选目标亚群时用的是hERV家族的平均表达量,而hERV与基因的相关性并不体现在“平均表达量”上
- 之前在相关论文中看到说hERVK与TLR8表达相关,想看看我这里是不是也是这个情况,如果能得到类似的结果,就试一试位点级的分析(毕竟开题时候提的就是位点级分析)
- 在目标亚群内部,计算hERVK和全部基因的相关性(不是前面挑出来的100多个基因),看高相关度的基因是否与AD状态有关、是否是这个亚群与其它亚群的差异表达基因,并进行GO富集
- GO富集时可去掉一些与“核糖体/线粒体”相关的基因,让结果尽可能在自噬/溶酶体、应激反应、mTOR/蛋白稳态、炎症/免疫相关、脂代谢/膜运输,而不是核糖体/能量代谢/翻译/呼吸作用上