使用我的GSE157827数据进行初步分析-4
other-other2026.3.11-2026.3.18研究进展
用管家基因来验证测序深度对表达量计数的影响
hk_genes <- c("ACTB", "GAPDH", "RPL13A", "RPLP0", "EEF1A1", "PPIA")
hk_genes <- intersect(hk_genes, rownames(seu))
meta <- seu@meta.data %>%
rownames_to_column("cell") %>%
select(cell, group, nCount_RNA) %>%
filter(group %in% c("NC", "AD")) %>%
mutate(
group = factor(group, levels = c("NC", "AD")),
log10_nCount_RNA = log10(nCount_RNA + 1)
)
cells_use <- meta$cell
expr_mat <- GetAssayData(seu, assay = "RNA", slot = "data")[hk_genes, cells_use, drop = FALSE]
df_long <- lapply(seq_along(hk_genes), function(i) {
data.frame(
cell = cells_use,
gene = hk_genes[i],
expr = as.numeric(expr_mat[i, ]),
stringsAsFactors = FALSE
)
}) %>%
bind_rows() %>%
left_join(meta, by = "cell")
stat_res <- df_long %>%
group_by(gene) %>%
summarise(
NC_median = median(expr[group == "NC"], na.rm = TRUE),
AD_median = median(expr[group == "AD"], na.rm = TRUE),
diff_median = AD_median - NC_median,
p_wilcox = wilcox.test(expr ~ group)$p.value,
rho_nCount = suppressWarnings(cor(expr, log10_nCount_RNA,
method = "spearman",
use = "complete.obs")),
.groups = "drop"
) %>%
mutate(
p_wilcox_adj = p.adjust(p_wilcox, method = "BH"),
celltype = "All"
) %>%
arrange(p_wilcox_adj)
stat_by_celltype <- df_long %>%
group_by(celltype, gene) %>%
summarise(
NC_median = median(expr[group == "NC"], na.rm = TRUE),
AD_median = median(expr[group == "AD"], na.rm = TRUE),
diff_median = AD_median - NC_median,
p_wilcox = tryCatch(wilcox.test(expr ~ group)$p.value, error = function(e) NA_real_),
rho_nCount = suppressWarnings(cor(expr, log10_nCount_RNA,
method = "spearman",
use = "complete.obs")),
.groups = "drop"
) %>%
group_by(celltype) %>%
mutate(p_wilcox_adj = p.adjust(p_wilcox, method = "BH")) %>%
ungroup()
write.csv(bind_rows(stat_res, stat_by_celltype), file = "C:\\Users\\17185\\Desktop\\hERV_calc\\GSE157827\\my_res\\ncount_gene.csv", row.names = F)
# AD和NC中这些管家基因的表达分布
ggplot(df_long, aes(x = group, y = expr, fill = group)) +
geom_violin(scale = "width", trim = TRUE) +
geom_boxplot(width = 0.12, outlier.size = 0.2, fill = "white") +
facet_wrap(~ gene, scales = "free_y", ncol = 3) +
stat_compare_means(method = "wilcox.test", label = "p.format") +
theme_classic(base_size = 12)
# 这些管家基因表达和nCount_RNA是否仍明显相关
ggplot(df_long, aes(x = log10_nCount_RNA, y = expr)) +
geom_point(size = 0.2, alpha = 0.25) +
geom_smooth(method = "lm", se = FALSE) +
facet_wrap(~ gene, scales = "free_y", ncol = 3) +
theme_classic(base_size = 12)
gtf文件-hERV家族名
其它AD数据汇总
继续之前的细胞类型-hERV家族水平重调分析
调整协变量后的新差异表达结果
总的来说,其实相差不是很大,而且变化的主要是p值
全体细胞的超家族:
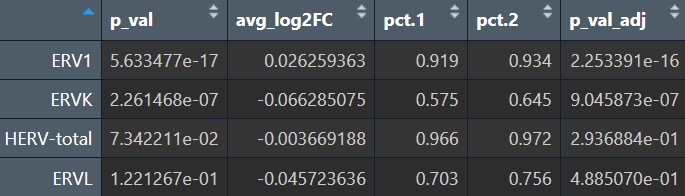
各细胞类型的超家族:
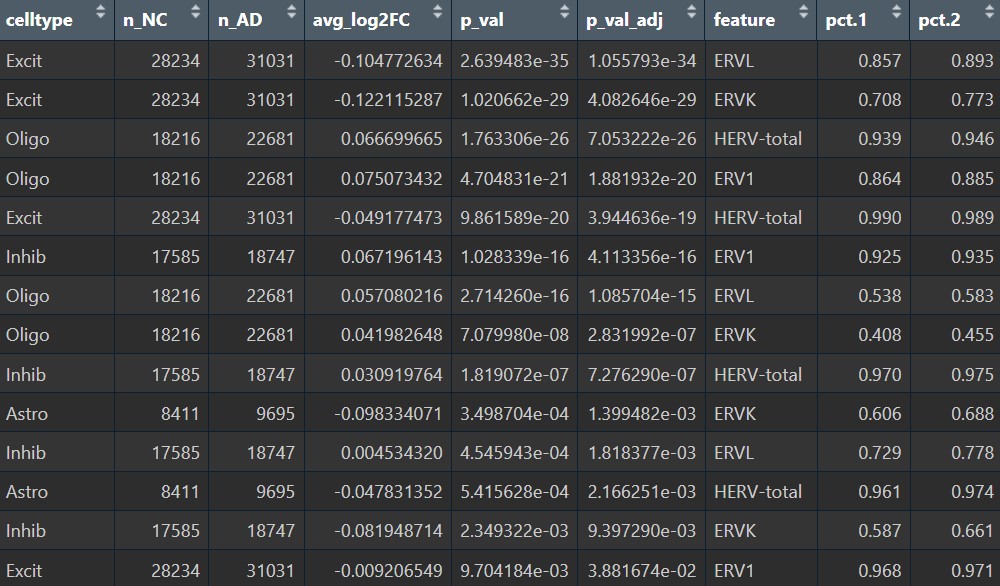
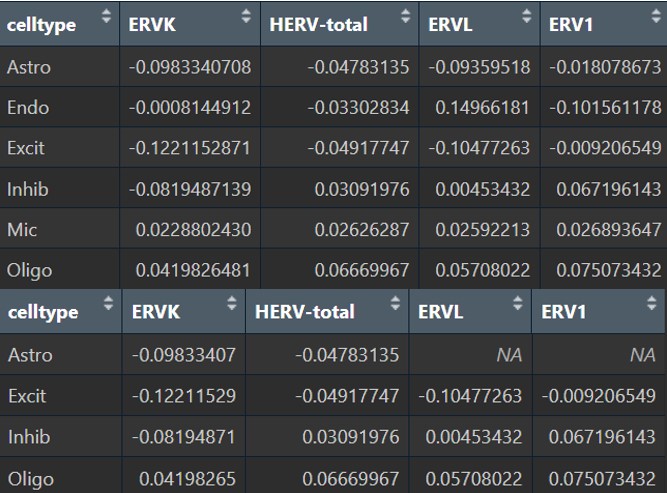
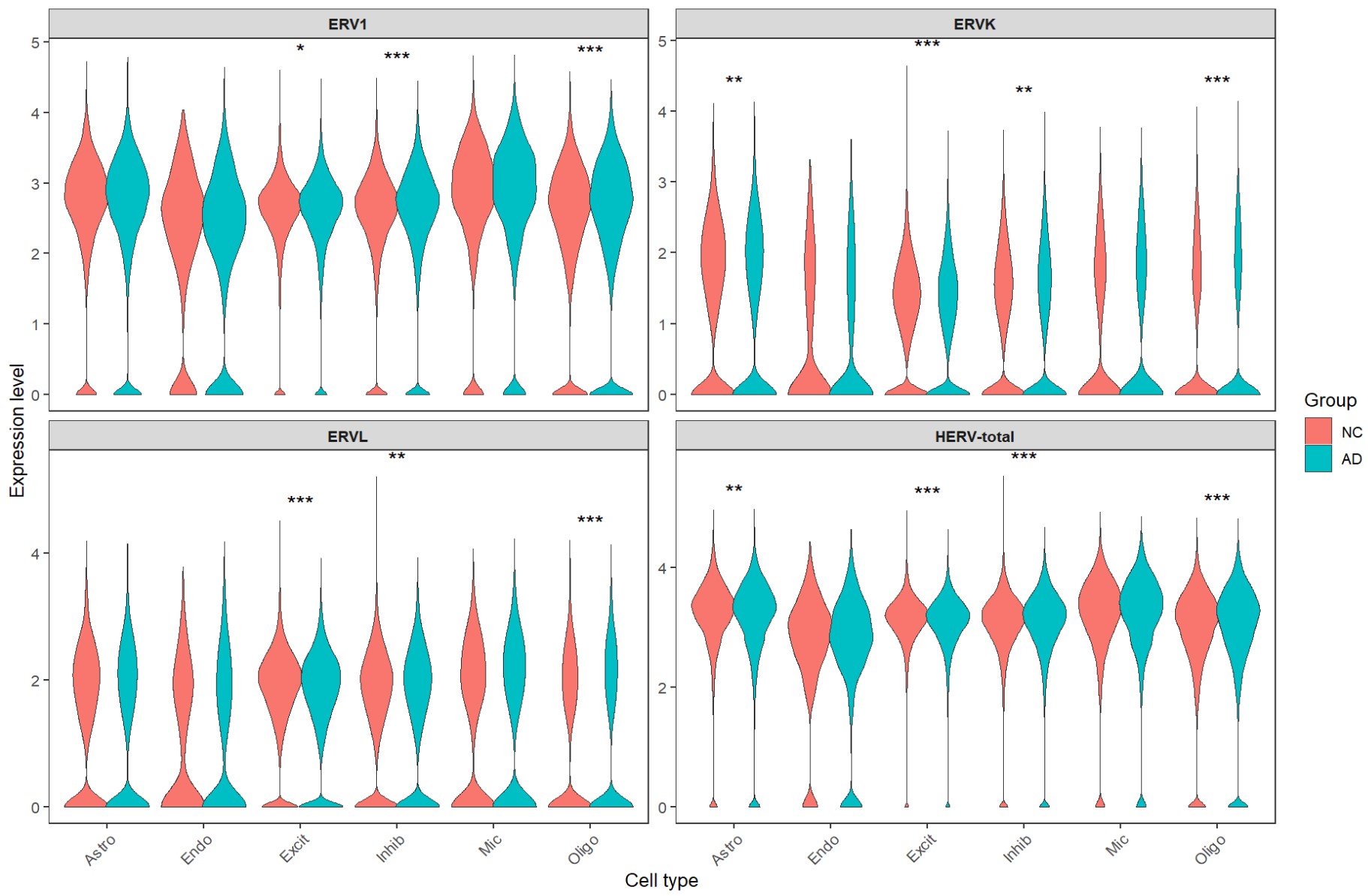
inhib类型在这步里变得比较显著,也许也可以作为分析对象之一
各细胞类型的亚家族:
这里的变化就稍微有点大了,比如astro的HERVK的p值就变成了1(而原来是<0.05的),然后HERV3这个家族看起来表达量很高的样子
调整oligo的分辨率再看看表达量热图
除了改分辨率之外,对于目标hERV家族的选取也尽可能宽松一些,防止有些hERV家族只在某些亚群中特异表达而被过滤掉
oligo$oligo_subcluster <- oligo$RNA_snn_res.0.1
# oligo$oligo_subcluster <- oligo$RNA_snn_res.0.3
Idents(oligo) <- oligo$oligo_subcluster
DimPlot(oligo, reduction = "umap", group.by = "oligo_subcluster", label = TRUE)
res_subfamily <- read.csv("C:\\Users\\17185\\Desktop\\hERV_calc\\GSE157827\\my_res\\hERV_family.csv")
res_oligo_focus <- res_subfamily %>%
filter(
celltype == "oligo",
pmax(pct.1, pct.2) >= 0.10, # 至少一组有一定检出
pmin(pct.1, pct.2) >= 0.02 # 另一组不要过于稀疏
) %>%
arrange(desc(abs(avg_log2FC)), p_val_adj)
down_subfam <- res_oligo_focus %>% # 下调
filter(avg_log2FC < 0) %>%
arrange(avg_log2FC) %>%
pull(subfamily)
up_subfam <- res_oligo_focus %>% # 上调
filter(avg_log2FC > 0) %>%
arrange(desc(avg_log2FC)) %>%
pull(subfamily)
# 画图代码不变



Oligo
oligo <- subset(seu, subset = celltype == "Oligo")
DefaultAssay(oligo) <- "RNA"
oligo <- FindVariableFeatures(
oligo,
selection.method = "vst",
nfeatures = 2000
)
oligo <- ScaleData(
oligo,
features = VariableFeatures(oligo),
vars.to.regress = c("nFeature_RNA", "percent_mito")
)
oligo <- RunPCA(
oligo,
features = VariableFeatures(oligo),
)
ElbowPlot(oligo, ndims = 50)

pc.num <- 1:30
oligo <- RunUMAP(
oligo,
dims = pc.num
)
DimPlot(oligo, group.by = "group") | DimPlot(oligo, split.by = "group")

oligo <- FindNeighbors(
oligo,
dims = pc.num
)
oligo <- FindClusters(
oligo,
resolution = c(0.01,0.05,0.1,0.2,0.3,0.4,0.5)
)
clustree(oligo@meta.data, prefix = "RNA_snn_res.")

oligo$oligo_subcluster <- oligo$RNA_snn_res.0.3
Idents(oligo) <- oligo$oligo_subcluster
DimPlot(oligo, reduction = "umap", group.by = "oligo_subcluster", label = TRUE)
saveRDS(
oligo,
file = "C:\\Users\\17185\\Desktop\\hERV_calc\\GSE157827\\my_data\\GSE157827_oligo.rds",
compress = "xz"
)
