使用我的GSE157827数据进行初步分析-5
other-other2026.3.18-2026.4.8研究进展
GSE174367
先看这几个1/2/3都是什么部分:
cd /public/home/GENE_proc/wth/GSE174367/fastq/
for fq in SRR14513977_{1,2,3}.fastq.gz; do
echo "===== $fq ====="
zcat "$fq" | awk '
NR%4==2 && ++n<=200000 {len[length($0)]++}
END{
for (l in len) print l, len[l]
}' | sort -n
echo
done
SRR14513977_1.fastq.gz:都是8bp,是sample index read- 正常来讲另外两个应该一个是28bp,另一个是90-100bp,但很神奇的是另外两个都是100bp
- GPT说是没切成28bp的形式,而是原始格式,需要自己看一下哪个是barcode+UMI,哪个是cDNA
import gzip
import sys
import math
from collections import Counter
N = 200000
MAX_POS = 40
def rc(seq):
comp = str.maketrans("ACGTN", "TGCAN")
return seq.translate(comp)[::-1]
def iter_seqs(fq, n=N):
count = 0
with gzip.open(fq, "rt") as f:
for i, line in enumerate(f):
if i % 4 == 1:
yield line.strip()
count += 1
if count >= n:
break
def entropy(counter):
total = sum(counter.values())
if total == 0:
return 0.0
h = 0.0
for v in counter.values():
p = v / total
h -= p * math.log2(p)
return h
def load_whitelist(path):
wl = set()
opener = gzip.open if path.endswith(".gz") else open
with opener(path, "rt") as f:
for line in f:
wl.add(line.strip())
return wl
def analyze_fastq(fq, whitelist=None):
seqs = list(iter_seqs(fq, N))
if not seqs:
print(f"\n===== {fq} =====")
print("No reads found")
return
lens = Counter(map(len, seqs))
dom_len, dom_n = lens.most_common(1)[0]
print(f"\n===== {fq} =====")
print(f"sampled reads: {len(seqs)}")
print("length distribution:")
for l, c in sorted(lens.items()):
print(f" {l} bp\t{c}")
print(f"dominant length: {dom_len} bp ({dom_n/len(seqs):.2%})")
p16 = [s[:16] for s in seqs if len(s) >= 16]
p28 = [s[:28] for s in seqs if len(s) >= 28]
c16 = Counter(p16)
c28 = Counter(p28)
print(f"unique first16: {len(c16)} / {len(p16)} ({len(c16)/len(p16):.2%})")
print(f"unique first28: {len(c28)} / {len(p28)} ({len(c28)/len(p28):.2%})")
print("top 10 first16:")
for s, c in c16.most_common(10):
print(f" {c}\t{s}")
print("top 10 first28:")
for s, c in c28.most_common(10):
print(f" {c}\t{s}")
if whitelist is not None:
hit = sum((x in whitelist) for x in p16)
hit_rc = sum((rc(x) in whitelist) for x in p16)
print(f"whitelist hit rate (first16): {hit/len(p16):.2%}")
print(f"whitelist hit rate (reverse-complement first16): {hit_rc/len(p16):.2%}")
tails = [s[28:60] for s in seqs if len(s) >= 60]
polyA = sum(("AAAAAAAAAAAA" in x) for x in tails)
polyT = sum(("TTTTTTTTTTTT" in x) for x in tails)
print(f"reads with >=12A in pos29-60: {polyA/len(tails):.2%}")
print(f"reads with >=12T in pos29-60: {polyT/len(tails):.2%}")
pos_counts = [Counter() for _ in range(MAX_POS)]
for s in seqs:
for i, b in enumerate(s[:MAX_POS]):
pos_counts[i][b] += 1
print("\nPer-cycle summary (first 40 cycles):")
print("pos\tmajor_base\tmajor_frac\tentropy\tA\tC\tG\tT\tN")
for i, cnt in enumerate(pos_counts, start=1):
total = sum(cnt.values())
if total == 0:
continue
major_base, major_n = cnt.most_common(1)[0]
A = cnt.get("A", 0) / total
C = cnt.get("C", 0) / total
G = cnt.get("G", 0) / total
T = cnt.get("T", 0) / total
Nn = cnt.get("N", 0) / total
H = entropy(cnt)
print(f"{i}\t{major_base}\t{major_n/total:.3f}\t{H:.3f}\t{A:.3f}\t{C:.3f}\t{G:.3f}\t{T:.3f}\t{Nn:.3f}")
if __name__ == "__main__":
if len(sys.argv) < 3:
print("Usage: python inspect_10x_cb_umi.py whitelist.txt.gz file1.fastq.gz file2.fastq.gz ...")
sys.exit(1)
wl = load_whitelist(sys.argv[1])
for fq in sys.argv[2:]:
analyze_fastq(fq, wl)
cd /public/home/GENE_proc/wth/GSE174367/fastq/
python ../inspect_10x_cb_umi.py \
/public/home/wangtianhao/Desktop/STAR_ref/whitelist/3M-february-2018.txt \
SRR14513977_2.fastq.gz \
SRR14513977_3.fastq.gz
最后算出来_2的前16bp对whitelist的命中率高达96%,并且它的unique first16只有14.52%,但加上12bp以后,unique first28达到91.62%,说明前16位是barcode,后12bp是UMI
先跑一个测试:
module load miniconda3/base
conda activate STAR
cd /public/home/GENE_proc/wth/GSE174367/
STAR \
--runMode alignReads \
--runThreadN 8 \
--genomeDir /public/home/wangtianhao/Desktop/STAR_ref/hg38/ \
--readFilesIn fastq/SRR14513977_3.fastq.gz fastq/SRR14513977_2.fastq.gz \
--readFilesCommand zcat \
--outFileNamePrefix star_test/SRR14513977 \
--soloType CB_UMI_Simple \
--soloCBstart 1 \
--soloCBlen 16 \
--soloUMIstart 17 \
--soloUMIlen 12 \
--soloBarcodeReadLength 0 \
--soloCBwhitelist /public/home/wangtianhao/Desktop/STAR_ref/whitelist/3M-february-2018.txt \
--soloFeatures GeneFull \
--clipAdapterType CellRanger4 \
--soloCellFilter EmptyDrops_CR \
--soloCBmatchWLtype 1MM_multi_Nbase_pseudocounts \
--soloUMIfiltering MultiGeneUMI_CR \
--soloUMIdedup 1MM_CR
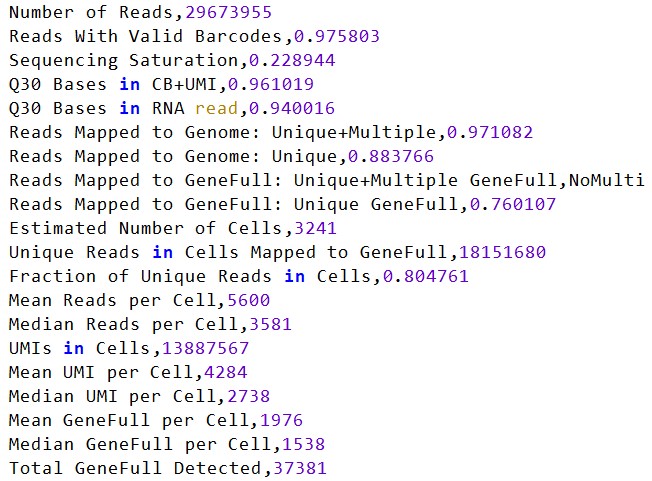
再跑一个完整的样本(包含多个SRR)
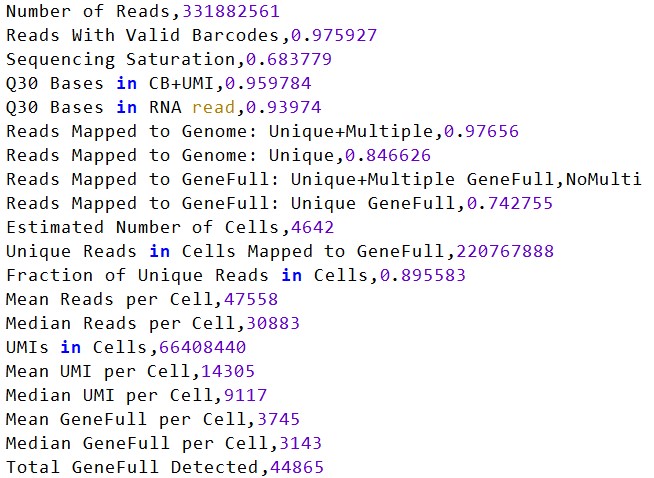
这个GSM5292838在作者的统计表里有4605个细胞(我的有4642个细胞),可以说是完全吻合,由此这个计数参数应该是没有什么问题的
总结:
- 6w多个细胞,来自18个样本,虽然GEO数据库中有19个,但根据论文的补充材料,作者只使用了其中的18个,实测可能是因为差的那个测序质量不太好(nCount_RNA比较低)
- 测序深度较深,nCount_RNA/nFeature_RNA较大(虽然细胞数只有前组数据的1/3,但fastq文件大小却接近3/4)
- 在该数据对应的论文中,作者分了7种细胞类型,在之前的6种基础上,增加了“少突胶质前体细胞”(OPC, oligodendrocyte progenitor cells)——具有分化生成成熟少突胶质细胞的能力,在大脑早期发育的髓鞘形成过程中起重要作用
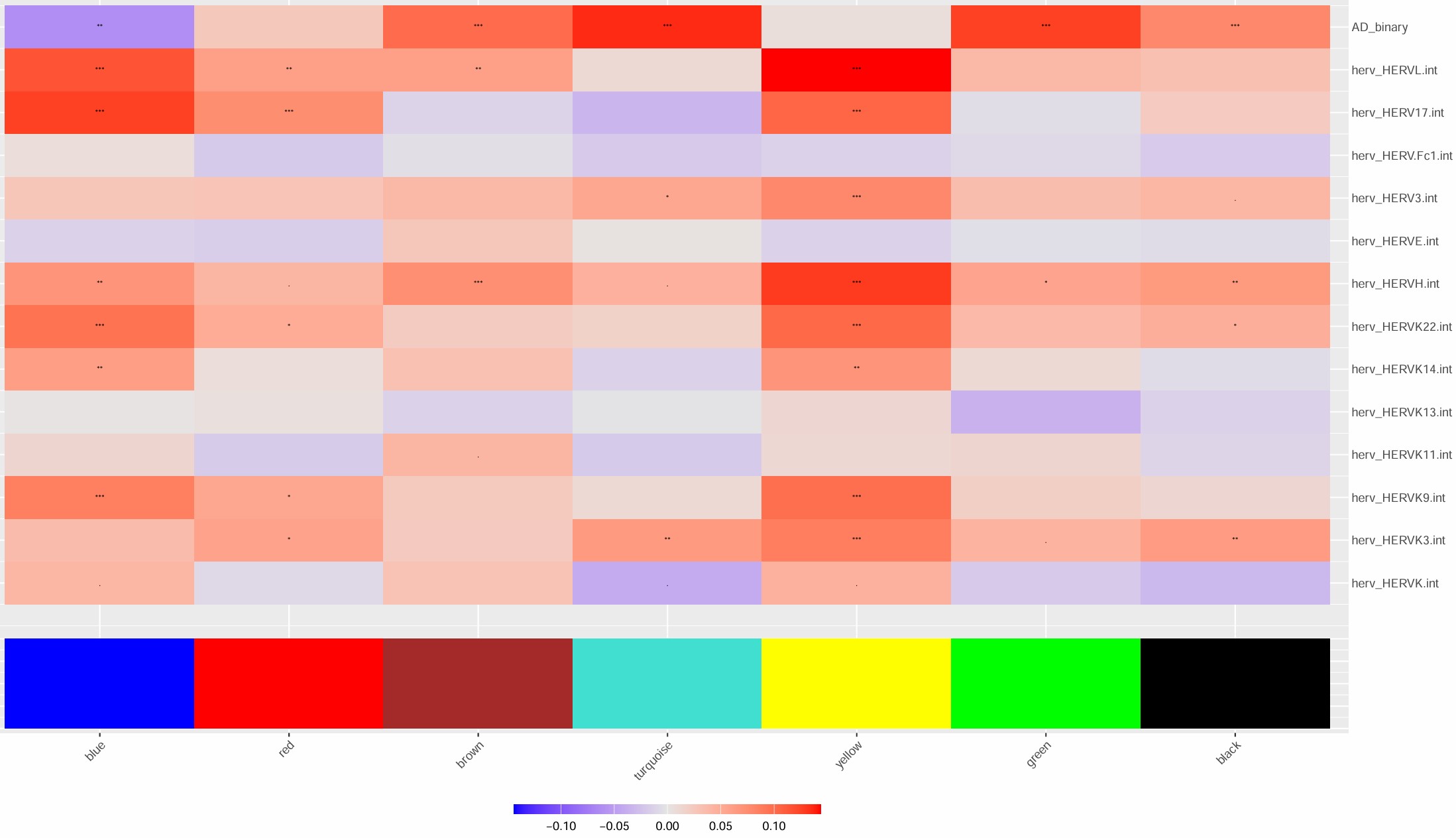
hERV ~ gene
主要思路:
- 在细胞类型层面做hERV~gene共表达分析(不在亚群层面上):构建共表达网络,把网络中邻近的基因进行功能富集
- 在各细胞类型里找hERV特异表达的亚群,聚焦到某些亚群中高表达的subfamilies做一些定性分析:看这个亚群中特异表达的hERV在共表达网络中连接的基因,对比这个亚群与其它亚群的差异表达基因,看是否与AD有关
- 验证hERVK与TLR8表达相关(先所有细胞,再看细胞类型),参照那篇论文,为下一步hERV位点分析选择位点?
共表达网络test
在之前的代码中,我做的是一个hERV和一个gene的相关性或线性拟合,现在想做一下网络层面的关系:
- 在某个大细胞类型里,哪些基因会一起变化、组成模块,这些模块和hERV是否相关
- 某个hERV高表达亚群里,究竟激活了哪一类网络程序
- WGCNA/hdWGCNA(用于单细胞数据的WGCNA):先用表达相关性构建加权网络,再把高度共表达的基因聚成模块,用模块特征值去和外部性状做关联
- 在构建完网络之后,可以在某个和hERV明显相关的模块里,再去找和这个hERV相关性最高的基因(一个hERV和一个gene的线性拟合)
方案1:先用基因构建共表达网络,再把hERV当作外部性状去挂到网络上(GPT推荐)
- 在细胞大类中,单独构建基因的共表达网络,得到若干个基因模块
- 选在该细胞类型中有一定表达比例的基因
- 把hERV的表达和模块特征值做关联
- 选定一些trait:hERV家族表达量、AD/NC等
- 模块特征值和trait的相关分析,画热图
- 找到和某个hERV家族显著相关的模块
- 例如某个模块和hERVK表达显著相关,同时和AD状态相关,那么这个模块就是一个很有价值的“hERV-AD关联模块”
- 后续对这些模块进行亚群级别的分析(具体方法在方案1中有讲)
方案2:把hERV当成普通基因作为网络节点直接参与建网
- 取网络中与hERV相邻的一些基因进行亚群级别的分析
- GPT不推荐,说是会存在一些问题:
- hERV家族数量远少于基因,且表达更稀疏
- hERV节点本身不能直接做GO
- 在WGCNA时,hERV要和普通基因一同作为feature进行标准化,这样就和之前单独标准化的思路不同,不过考虑到hERV表达量很低,影响也许不大?
采用哪种方案比较好:
- 方案1会更好解释家族水平hERV
- 方案2看起来更适合探讨位点级别的局部网络
共表达网络的5个对象:
- 节点(node):通常是gene
- 边(edge):表示两个基因在同一大细胞类型内、在不同样本/细胞中的表达是否同步变化
- 模块(module):一群彼此更紧密共表达的基因,往往对应某种生物过程
- 模块特征值(module eigengene, ME):本质上是模块表达矩阵的一个总结量,可以理解成“这个模块整体表达水平的代表值”,用于回答“这个模块在AD中是否表达更高”等问题
- hub gene:模块里连接度高、最核心的基因,通常是模块的代表基因或关键解释点。hdWGCNA里常用kME去衡量这种模块内连接性
共表达网络其它的一些专业名词:
- metacell:把若干个彼此相近、而且来自同一分组里的单细胞做平均,得到一个更稳定的小样本单元,如果设置为
group.by = c("wgcna_celltype", "orig.ident")就是先限制在同一个大细胞类型里,再限制在同一个sample里,然后在这些细胞里找相近细胞做平均。既保留了细胞类型特异性,又降低了单细胞水平分析的噪声 - trait:和模块做关联的外部变量——某个module的整体表达水平,是否随着某个trait变化
- soft power:WGCNA不会直接把基因之间的相关性(0.2/0.5/0.8这种)当网络中边的值,而是会加一个指数soft power,决定要把“高相关”和“低相关”的差距拉大到什么程度。这个指数变换可以把弱相关压小、强相关放大,如果power过小,网络对强弱相关的区分就不够;power慢慢增大,使网络结构更接近WGCNA想要的“scale-free”特征;如果power过大,整体网络会越来越稀,很多真实联系可能被压掉
- scale-free:无标度,指的是一种网络结构——大多数节点连接很少,只有少数节点连接特别多,也就是网络里会有少量枢纽节点(hub gene),而不是每个节点都连得差不多
- TOM:拓扑重叠矩阵(Topological Overlap Matrix),不是只看两个基因自己相关不相关,而是进一步看它们是不是拥有很多共同邻居
- 基因A和基因B直接相关,当然说明它们可能有关
- 但如果A和B不仅彼此相关,而且还都和C、D、E一起相关,那说明它们在网络中的“位置”也很像
- 这种“共同邻居很多”的关系,TOM会给更高分
所以它比单纯相关性更稳一些,因为它用了局部网络结构信息,而不是只看一条边
方案1
构建纯gene的共表达网络:
herv_assay <- "HERV_family"
celltype_name <- "Oligo"
herv_traits <- c("HERV3-int", "HERVH-int", "HERVE-int", "HERVK-int", "HERVK3-int", "HERVK9-int", "HERVK11-int", "HERVK13-int", "HERVK14-int", "HERVK22-int")
DefaultAssay(seu) <- "RNA"
seu$AD_binary <- ifelse(seu$group == "AD", 1, 0)
# 把trait写入metadata
mat <- LayerData(seu, assay = herv_assay, layer = "data")[herv_traits, , drop = FALSE]
mat <- t(as.matrix(mat))
colnames(mat) <- paste0("herv_", make.names(herv_traits))
seu <- AddMetaData(seu, metadata = as.data.frame(mat))
# SetupForWGCNA
seu <- SetupForWGCNA( # 运行时间几分钟
seu,
gene_select = "fraction",
fraction = 0.05, # 过滤掉表达比例<5%的基因
assay = "RNA",
wgcna_name = "gene_net"
)
# 构建metacell(按样本分开)
seu <- MetacellsByGroups( # 运行时间20min,需要10G+内存
seurat_obj = seu,
group.by = c("celltype", "orig.ident"),
ident.group = "celltype",
assay = "RNA",
layer = "data",
reduction = "pca",
dims = 1:30,
k = 25,
max_shared = 10,
min_cells = 50,
mode = "average",
wgcna_name = "gene_net"
)
seu <- NormalizeMetacells(seu, wgcna_name = "gene_net")
# 设置网络表达矩阵
seu <- SetDatExpr( # 运行时间几分钟
seu,
group_name = celltype_name,
group.by = "celltype",
use_metacells = TRUE,
assay = "RNA",
layer = "data",
wgcna_name = "gene_net"
)
# 选soft power
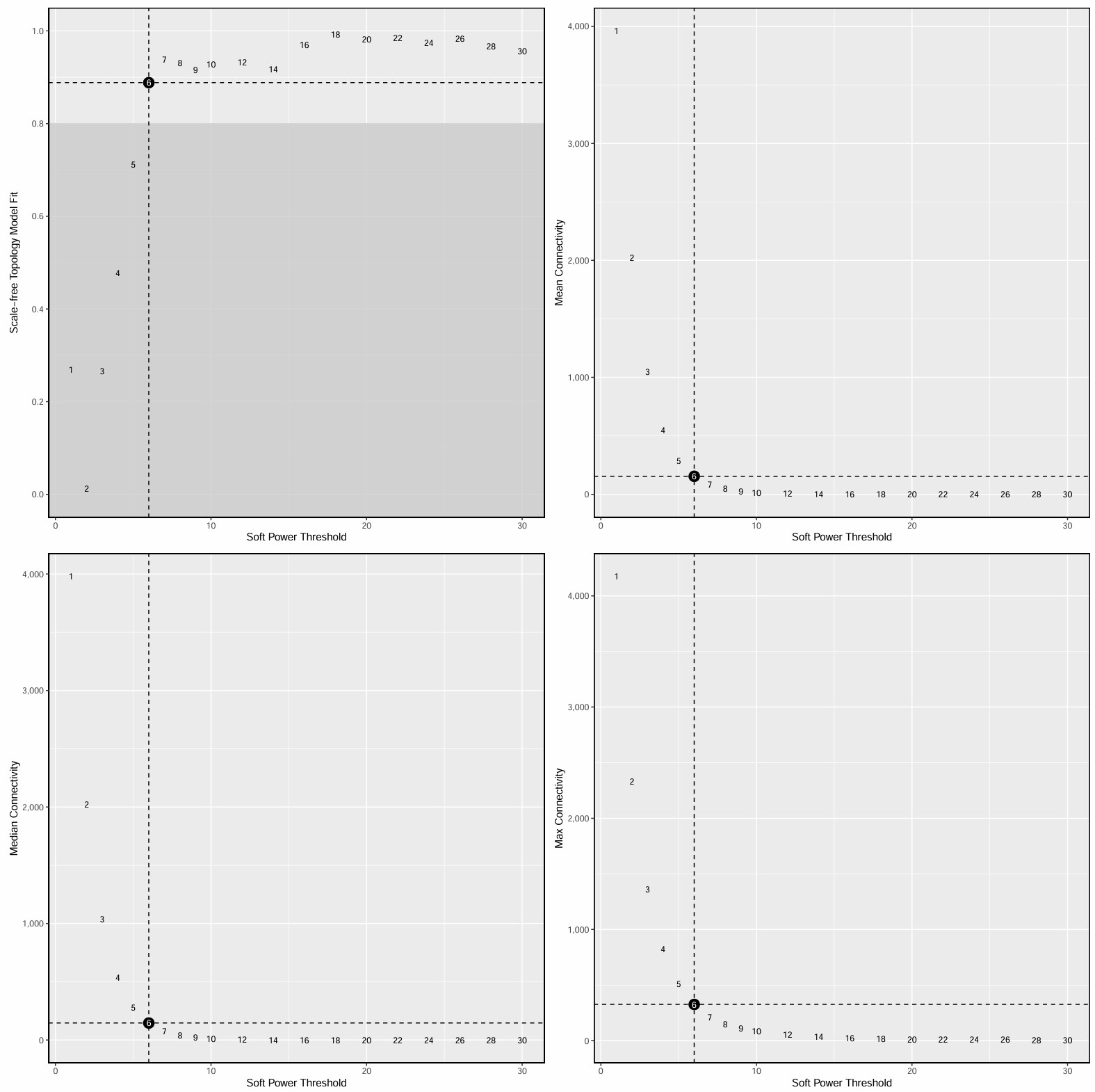
seu <- TestSoftPowers( # 运行时间几分钟
seu,
networkType = "signed",
wgcna_name = "gene_net"
)
power_table <- GetPowerTable(seu)
soft_power <- power_table$Power[which(power_table$SFT.R.sq >= 0.8)[1]]
if (is.na(soft_power)) {
soft_power <- power_table$Power[which.max(power_table$SFT.R.sq)]
}
plot_list <- PlotSoftPowers(seu)
pdf(file.path(res_dir, "oligo_soft_power.pdf"), width = 16, height = 16)
p <- patchwork::wrap_plots(plot_list, ncol = 2)
print(p)
dev.off()
save(seu, soft_power, herv_traits, file = file.path(res_dir, "oligo_WGCNA_before_ConstructNetwork.RData"))
# 构建网络
seu <- ConstructNetwork( # 运行时间20min,需要50G+内存
seu,
soft_power = soft_power,
networkType = "signed",
tom_name = "seu_TOM",
wgcna_name = "gene_net"
)
# 计算module eigengenes
seu <- ModuleEigengenes( # 运行时间20min,需要50G+内存
seu,
group.by.vars = "orig.ident",
assay = "RNA",
wgcna_name = "gene_net"
)
# module~trait关联
trait_cols <- c(
paste0("herv_", make.names(herv_traits)),
"AD_binary"
)
seu$celltype <- factor(seu$celltype)
seu <- ModuleTraitCorrelation(
seu,
traits = trait_cols,
group.by = "celltype",
wgcna_name = "gene_net"
)
mt <- GetModuleTraitCorrelation(seu)
# 结果
View(mt$cor) # 相关系数
View(mt$fdr) # FDR校正后p值
pdf(file.path(res_dir, "oligo_trait_corr.pdf"), width = 16, height = 16)
p <- PlotModuleTraitCorrelation(seu, label = "fdr", label_symbol = "stars")
print(p)
dev.off()
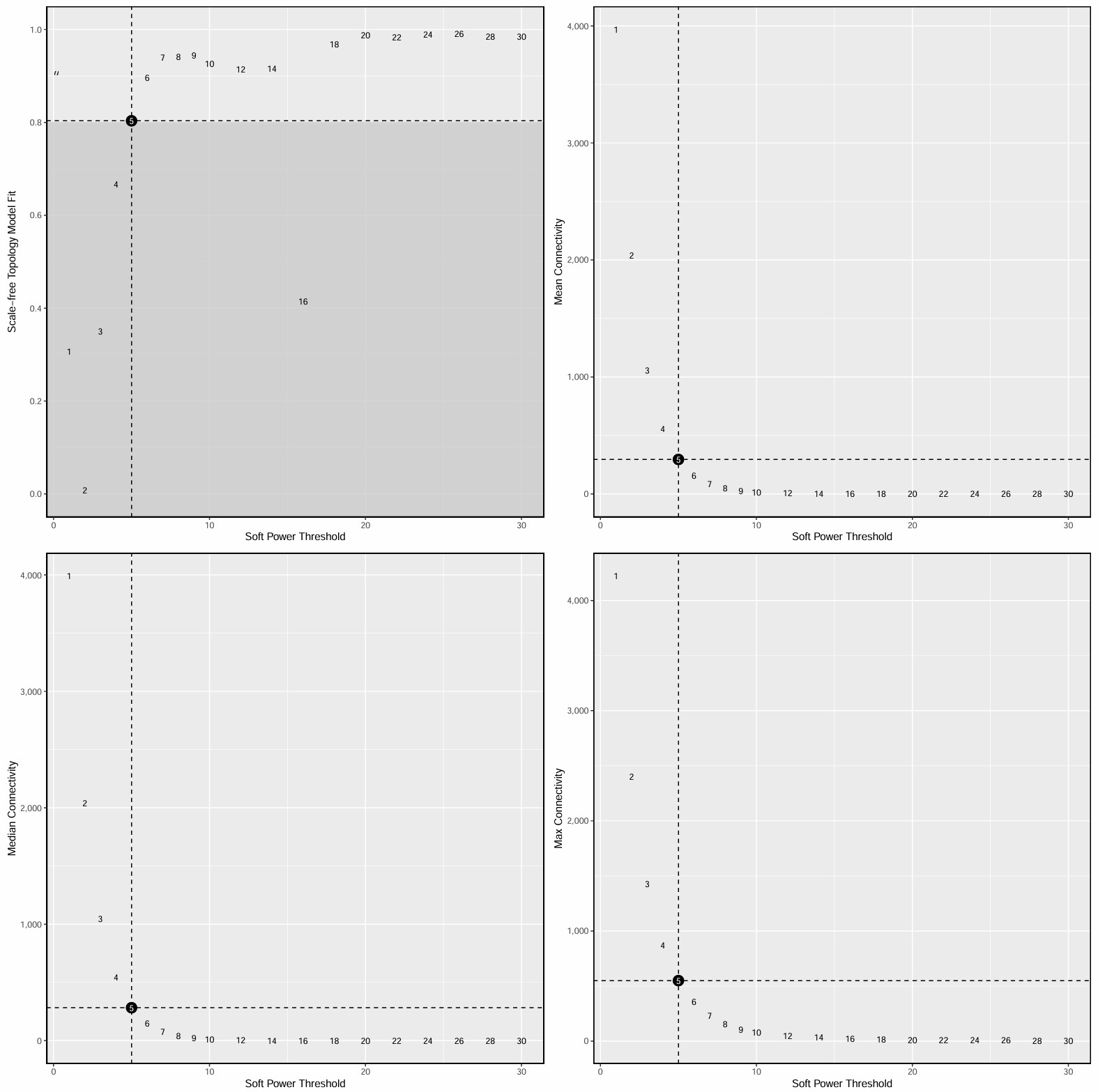
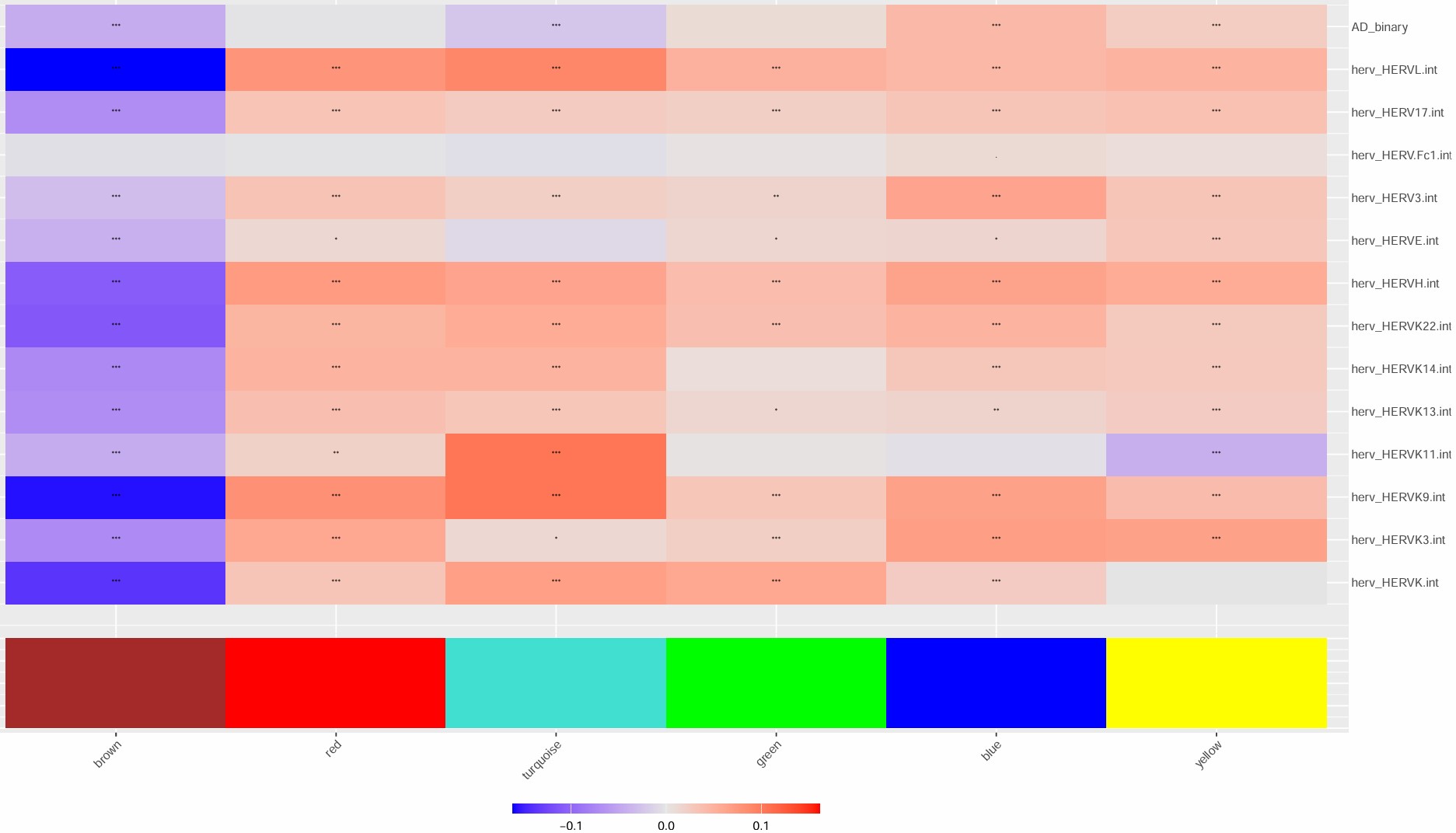
- 横轴是power,纵轴是scale-free topology model fit(signed R²),常取第一个让R²达到设定阈值(比如0.8左右)的power。如果数据本身比较噪、样本数少、或者是mixed network,达不到理想值也很常见,此时就选一个相对合理、且曲线已经进入平台区的power
- 横轴是power,纵轴是平均连接度(Mean connectivity),在“结构合理”和“不要过稀”之间找平衡
- 在这个结果中,power=5是一个比较合适的折中点,
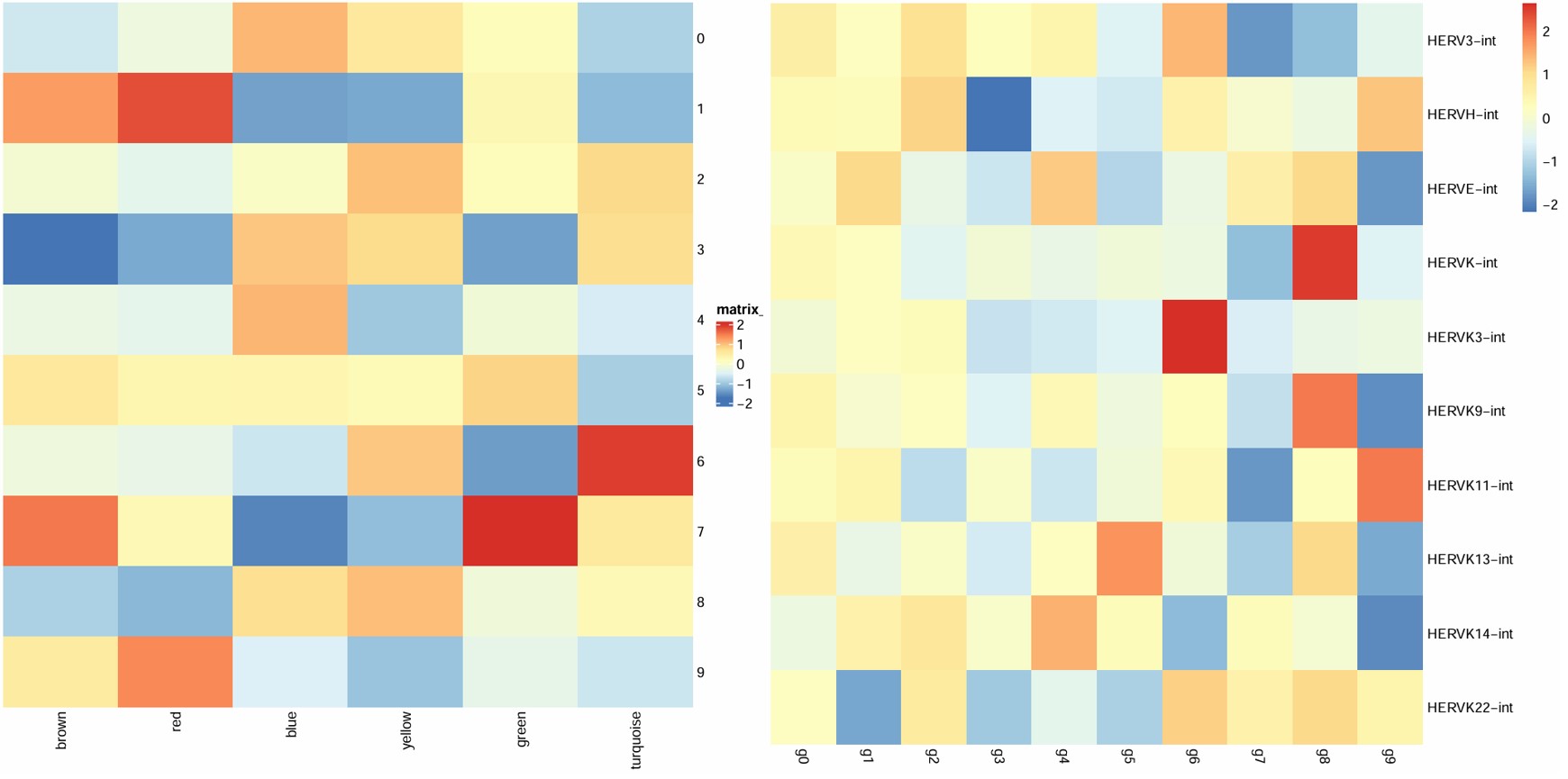
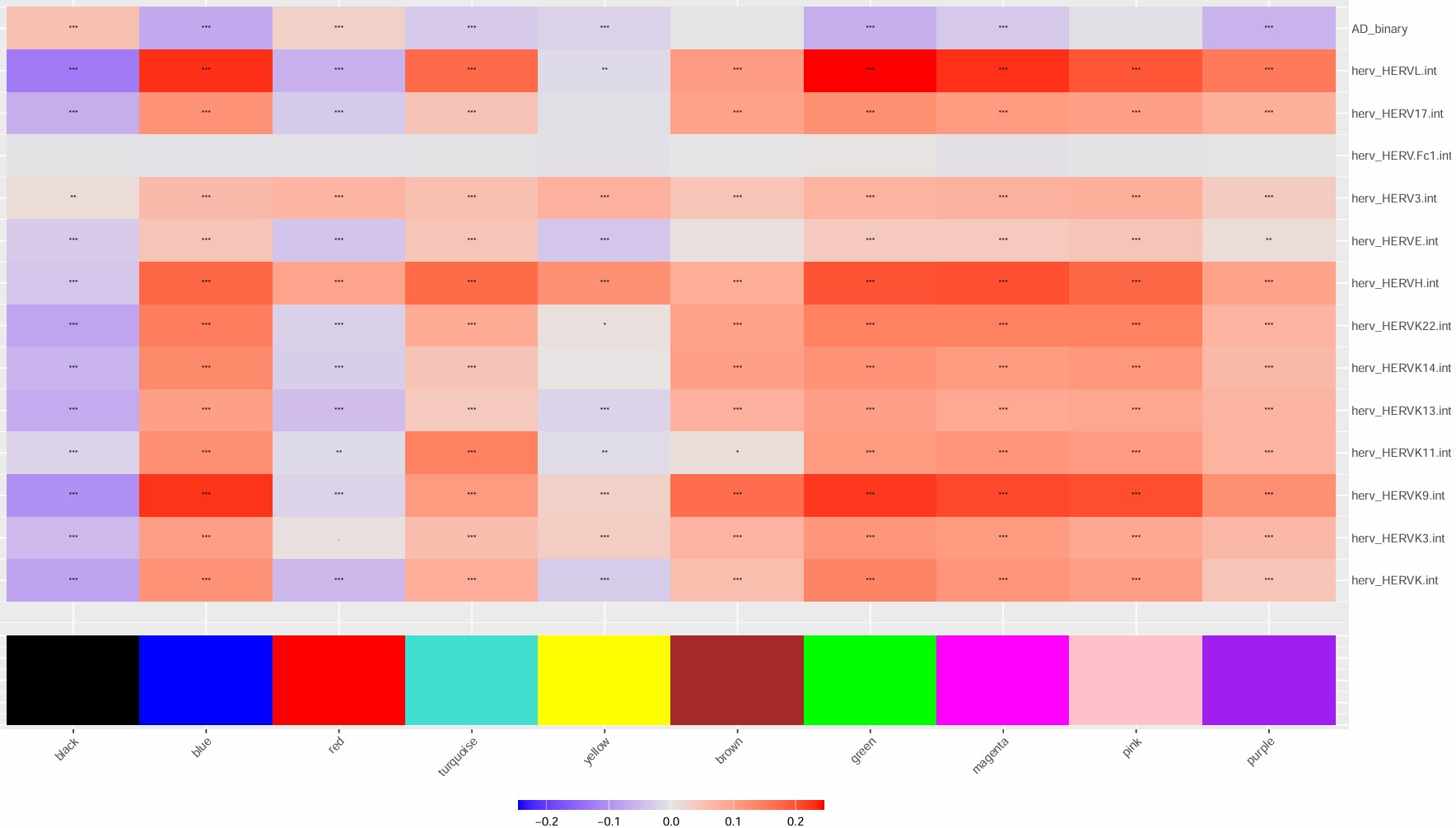
SFT.R.sq已达到阈值,连通性还保留得可以
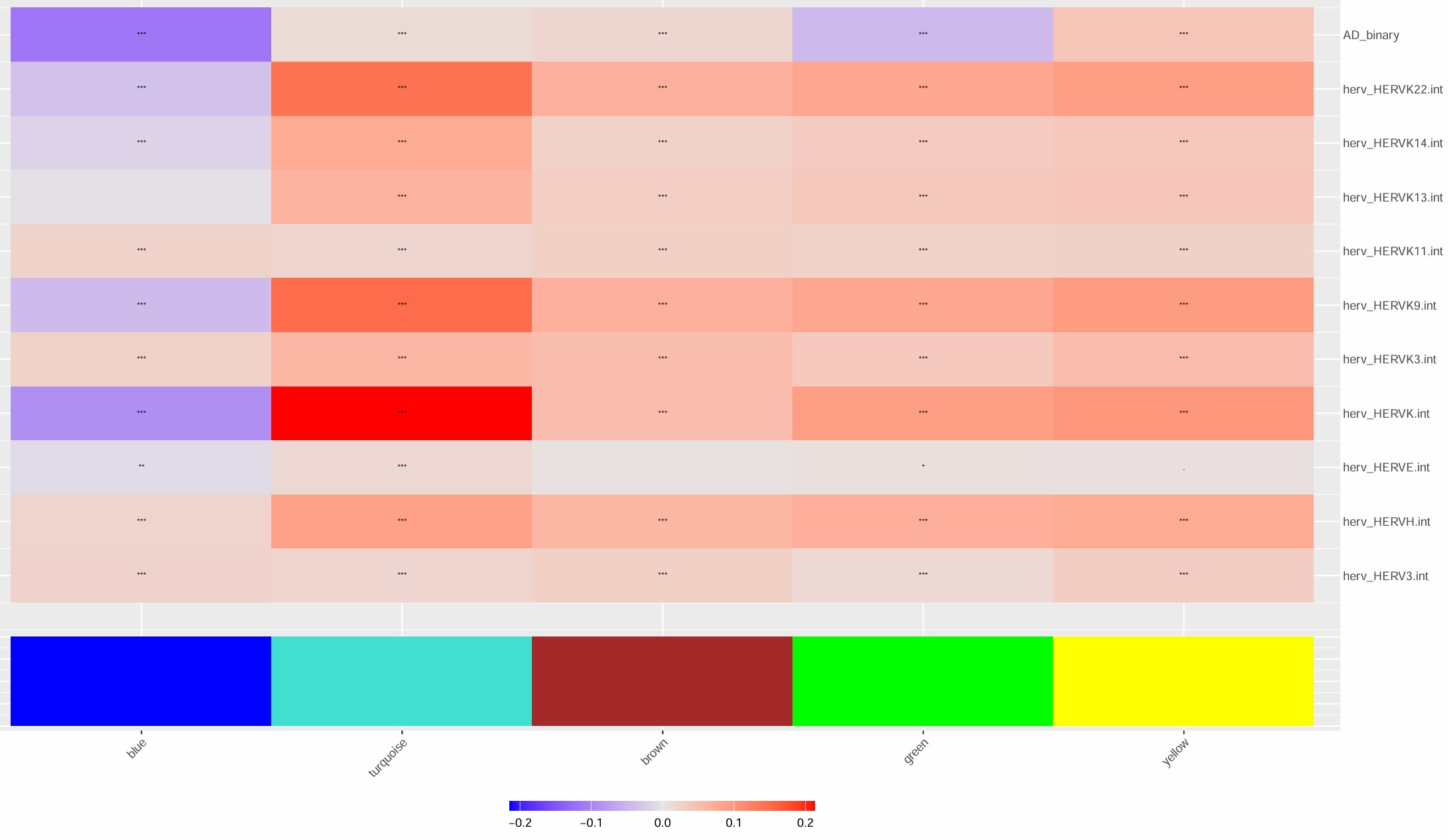
- turquoise模块最值得关注,几乎所有的hERV都是正相关
- blue模块和很多hERV是反向的
- 比较意外的是,这几个模块和AD的相关性竟然不是很高
- 不过总体来说相关度不算特别大,几乎都是-0.2~0.2的水平,
从gene network里提取“和某个hERV trait相关的模块”和hub genes
# 计算kME/hub genes
seu <- ModuleConnectivity(
seu,
group.by = "celltype",
group_name = celltype_name,
assay = "RNA",
wgcna_name = "gene_net"
)
save(seu, file = file.path(res_dir, "oligo_WGCNA.RData"))
# 目标hERV
trait_keep <- grep("^herv_", rownames(GetModuleTraitCorrelation(seu)$cor$all_cells), value = TRUE)
n_hubs <- 50 # 每个模块取多少个hub genes
pick_col <- function(df, candidates) {
x <- intersect(candidates, colnames(df))
if (length(x) == 0) stop("找不到列: ", paste(candidates, collapse = ", "))
x[1]
}
mat_to_df <- function(mat, value_name) {
df <- as.data.frame(as.table(as.matrix(mat)), stringsAsFactors = FALSE)
colnames(df) <- c("module", "trait", value_name)
df
}
# 选目标模块
fdr_cutoff <- 0.05 # 显著性阈值
cor_cutoff <- 0.05
mt <- GetModuleTraitCorrelation(seu)
cor_df <- mat_to_df(mt$cor$all_cells, "cor")
fdr_df <- mat_to_df(mt$fdr$all_cells, "fdr")
sig_tbl <- left_join(cor_df, fdr_df, by = c("module", "trait")) %>%
# filter(
# trait %in% trait_keep,
# module != "grey",
# !is.na(fdr),
# fdr < fdr_cutoff,
# abs(cor) >= cor_cutoff
# ) %>%
arrange(trait, desc(abs(cor)))
# write.csv(sig_tbl, file.path(res_dir, "oligo_module_trait_significant.csv"), row.names = FALSE)
# target_modules <- unique(sig_tbl$module)
target_modules <- c("turquoise", "blue") # 手动
# 提取模块基因 + hub genes
modules_df <- GetModules(seu)
hub_df <- GetHubGenes(seu, n_hubs = n_hubs)
gene_col_mod <- pick_col(modules_df, c("gene_name", "gene", "feature"))
module_col_mod <- pick_col(modules_df, c("module", "color"))
gene_col_hub <- pick_col(hub_df, c("gene_name", "gene", "feature"))
module_col_hub <- pick_col(hub_df, c("module", "color"))
# 背景基因设置为网络中的所有基因
universe_genes <- unique(modules_df[[gene_col_mod]])
universe_genes <- universe_genes[!is.na(universe_genes)]
# 只保留显著模块
module_gene_list <- lapply(target_modules, function(m) {
unique(modules_df[modules_df[[module_col_mod]] == m, gene_col_mod, drop = TRUE])
})
names(module_gene_list) <- target_modules
module_hub_list <- lapply(target_modules, function(m) {
unique(hub_df[hub_df[[module_col_hub]] == m, gene_col_hub, drop = TRUE])
})
names(module_hub_list) <- target_modules
# 导出模块基因和hub genes
# for (m in target_modules) {
# write.csv(
# data.frame(gene = module_gene_list[[m]]),
# file.path(res_dir, paste0("oligo_genes_", m, ".csv")),
# row.names = FALSE
# )
# write.csv(
# data.frame(hub_gene = module_hub_list[[m]]),
# file.path(res_dir, paste0("oligo_hub_genes_", m, ".csv")),
# row.names = FALSE
# )
# }
# 汇总结果
module_summary <- data.frame(
module = target_modules,
n_genes = sapply(module_gene_list, length),
n_hubs = sapply(module_hub_list, length),
top_hubs = sapply(module_hub_list, function(x) paste(head(x, 10), collapse = ", ")),
stringsAsFactors = FALSE
)
# write.csv(module_summary, file.path(res_dir, "oligo_module_summary.csv"), row.names = FALSE)
save(target_modules, universe_genes, module_gene_list, sig_tbl, module_hub_list, file = file.path(res_dir, "oligo_target_modules.RData"))
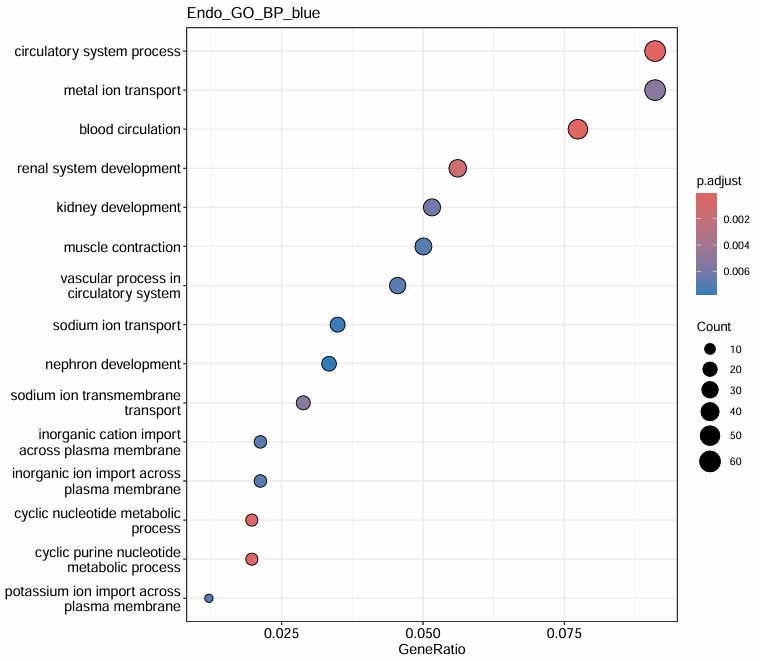
最后发现turquoise有3k多个基因,而blue只有300多个
# GO富集
run_go_one <- function(gene_vec, universe_genes = NULL, ont = "BP") {
gene_vec <- unique(na.omit(gene_vec))
if (length(gene_vec) < 10) return(NULL)
ego <- enrichGO(
gene = gene_vec,
universe = universe_genes,
OrgDb = org.Hs.eg.db,
keyType = "SYMBOL",
ont = ont,
pAdjustMethod = "BH",
pvalueCutoff = 1,
qvalueCutoff = 1,
readable = TRUE
)
if (is.null(ego) || nrow(as.data.frame(ego)) == 0) return(NULL)
ego
}
go_list <- lapply(target_modules, function(m) {
run_go_one(
gene_vec = module_gene_list[[m]],
universe_genes = universe_genes,
ont = "BP"
)
})
names(go_list) <- target_modules
for (m in target_modules) {
ego <- go_list[[m]]
if (is.null(ego)) next
go_df <- as.data.frame(ego)
write.csv(
go_df,
file.path(res_dir, paste0("GO_BP_", m, ".csv")),
row.names = FALSE
)
}
res <- list()
# 画图展示结果
for (m in target_modules) {
ego <- go_list[[m]]
if (is.null(ego)) next
res[[m]] <- dotplot(ego, showCategory = 15) +
ggtitle(paste0("GO BP - ", m))
}
pdf(file.path(res_dir, "oligo_module_trait_GO_summary.pdf"), width = 15, height = 24)
p <- wrap_plots(res, ncol = 2)
print(p)
dev.off()
# 汇总GO结果
top_go_tbl <- lapply(target_modules, function(m) {
ego <- go_list[[m]]
if (is.null(ego)) return(NULL)
go_df <- as.data.frame(ego)
go_df <- go_df[order(go_df$p.adjust), ]
trait_df <- sig_tbl %>%
filter(trait == m) %>%
arrange(desc(abs(cor)))
data.frame(
module = m,
trait = paste(trait_df$module, collapse = "; "),
cor = paste(round(trait_df$cor, 3), collapse = "; "),
n_gene = length(module_gene_list[[m]]),
hub_gene = paste(module_hub_list[[m]], collapse = "; "),
GO = paste(go_df$Description[1:20], collapse = "; "),
GO_padj = paste(go_df$p.adjust[1:20], collapse = "; "),
stringsAsFactors = FALSE
)
}) %>% bind_rows()
write.csv(top_go_tbl, file.path(res_dir, "oligo_module_trait_GO_summary2.csv"), row.names = FALSE)
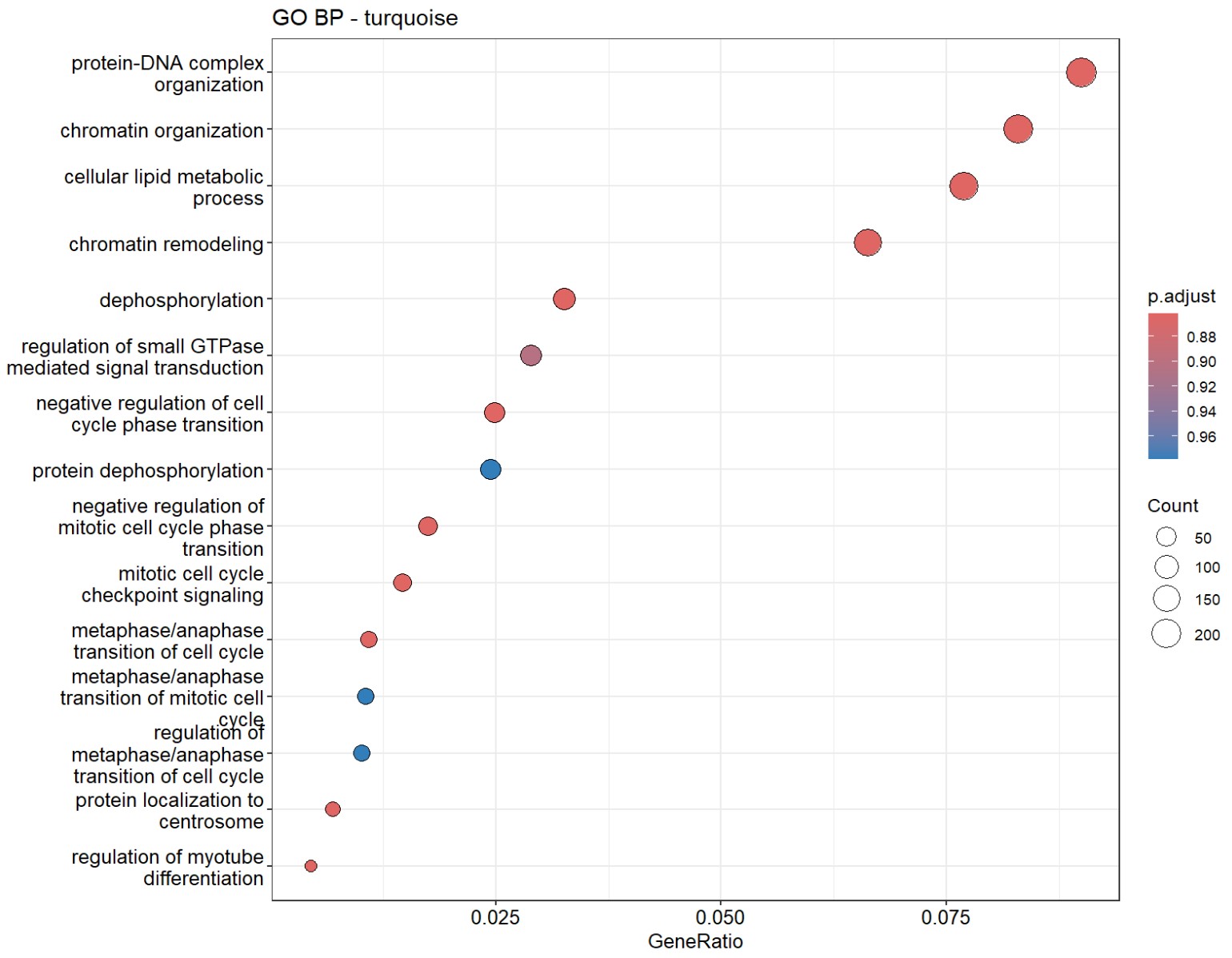
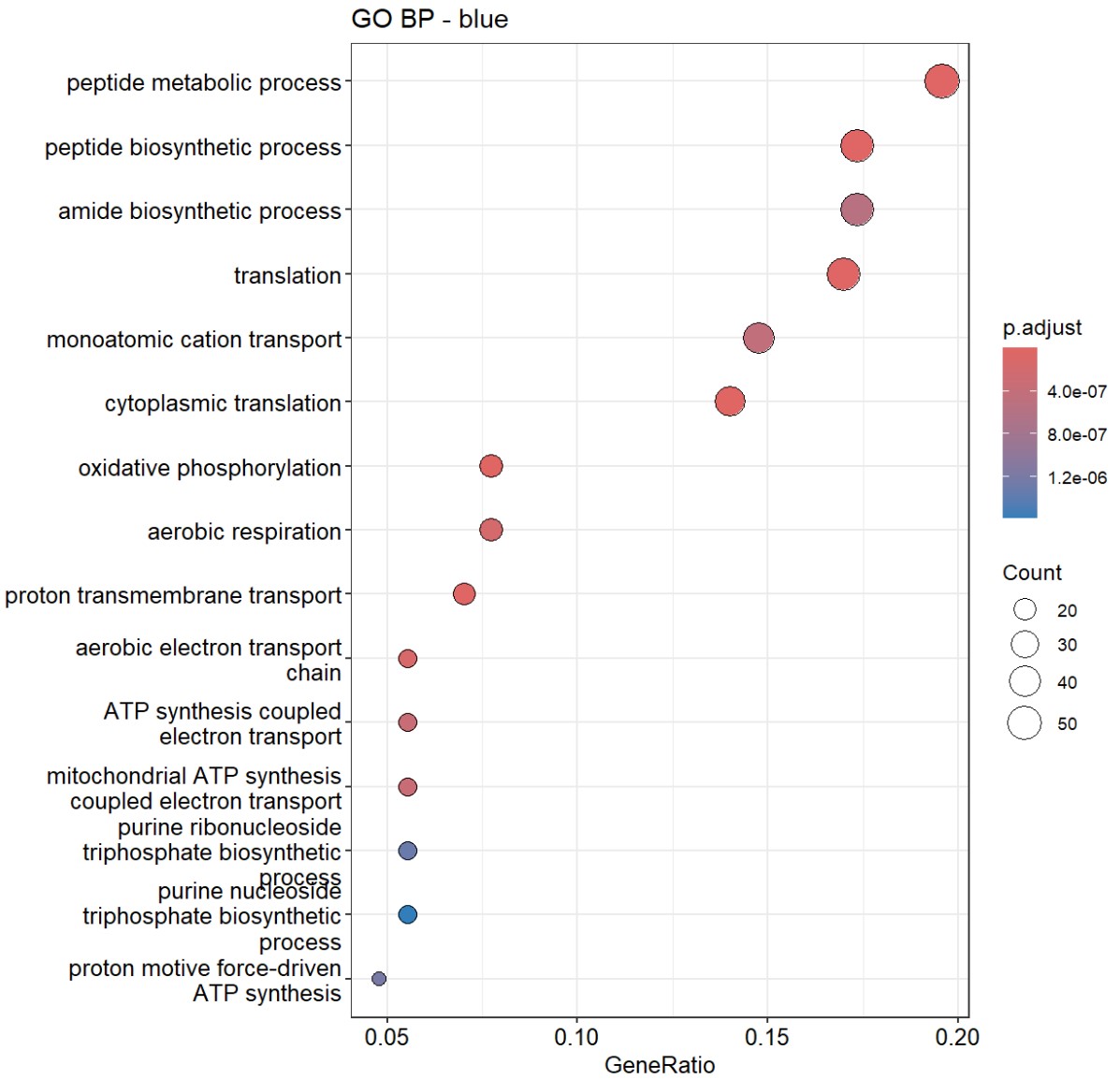
因为turquoise的基因数太多,所以p值就很高。又把两个模块的基因让GPT分析了一下,GPT也觉得turquoise更偏向总体细胞状态,blue的功能同质性更高:
- CNP/OLIG1/NKX6-2/HSPA2/APOD:少突成熟/髓鞘相关
- FTH1/FTL/CRYAB/SELENOP:代谢、应激、铁稳态/保护反应
- 有很多核糖体、线粒体呼吸链相关基因
说明blue很可能对应的是一种代谢活跃/翻译活跃/线粒体活跃/相对成熟稳态的少突程序,而AD和hERV又是负相关,说明在AD或hERV较高的状态下,这套“代谢-翻译-氧化磷酸化”程序是下降的
试一试改参数让模块更细一些?改ConstructNetwork中的两个参数:
fraction = 0.1:过滤掉更多的基因minModuleSize = 20:允许更小的模块保留下来mergeCutHeight = 0.05:减少相似模块合并
herv_assay <- "HERV_family"
celltype_name <- "Oligo"
herv_traits <- c("HERV3-int", "HERVH-int", "HERVE-int", "HERVK-int", "HERVK3-int", "HERVK9-int", "HERVK11-int", "HERVK13-int", "HERVK14-int", "HERVK22-int")
DefaultAssay(seu) <- "RNA"
seu$AD_binary <- ifelse(seu$group == "AD", 1, 0)
mat <- LayerData(seu, assay = herv_assay, layer = "data")[herv_traits, , drop = FALSE]
mat <- t(as.matrix(mat))
colnames(mat) <- paste0("herv_", make.names(herv_traits))
seu <- AddMetaData(seu, metadata = as.data.frame(mat))
seu <- SetupForWGCNA(
seu,
gene_select = "fraction",
fraction = 0.1,
assay = "RNA",
wgcna_name = "gene_net"
)
seu <- MetacellsByGroups(
seurat_obj = seu,
group.by = c("celltype", "orig.ident"),
ident.group = "celltype",
assay = "RNA",
layer = "data",
reduction = "pca",
dims = 1:30,
k = 25,
max_shared = 10,
min_cells = 50,
mode = "average",
wgcna_name = "gene_net"
)
seu <- NormalizeMetacells(seu, wgcna_name = "gene_net")
seu <- SetDatExpr(
seu,
group_name = celltype_name,
group.by = "celltype",
use_metacells = TRUE,
assay = "RNA",
layer = "data",
wgcna_name = "gene_net"
)
seu <- TestSoftPowers(
seu,
networkType = "signed",
wgcna_name = "gene_net"
)
power_table <- GetPowerTable(seu)
soft_power <- power_table$Power[which(power_table$SFT.R.sq >= 0.8)[1]]
if (is.na(soft_power)) {
soft_power <- power_table$Power[which.max(power_table$SFT.R.sq)]
}
seu <- ConstructNetwork(
seu,
soft_power = soft_power,
networkType = "signed",
overwrite_tom = TRUE,
tom_name = "seu_TOM",
deepSplit = 4,
minModuleSize = 20,
mergeCutHeight = 0.05,
pamStage = FALSE,
wgcna_name = "gene_net"
)
seu <- ModuleEigengenes(
seu,
group.by.vars = "orig.ident",
assay = "RNA",
wgcna_name = "gene_net"
)
trait_cols <- c(
paste0("herv_", make.names(herv_traits)),
"AD_binary"
)
seu <- ModuleTraitCorrelation(
seu,
traits = trait_cols,
group.by = "celltype",
wgcna_name = "gene_net"
)
get_module_size <- function(seu, wgcna_name) { # 看模块大小
df <- GetModules(seu, wgcna_name = wgcna_name)
gene_col <- intersect(c("gene_name", "gene", "feature"), colnames(df))[1]
mod_col <- intersect(c("module", "color"), colnames(df))[1]
out <- as.data.frame(table(df[[mod_col]]), stringsAsFactors = FALSE)
colnames(out) <- c("module", "n_genes")
out <- out[order(out$n_genes, decreasing = TRUE), ]
out
}
get_module_size(seu, "gene_net")
pdf(file.path(res_dir, "oligo_trait_corr.pdf"), width = 16, height = 16)
p <- PlotModuleTraitCorrelation(seu, label = "fdr", label_symbol = "stars")
print(p)
dev.off()
save(seu, file = file.path(res_dir, "oligo_WGCNA_try2.RData"))
module n_genes
3 grey 3157
2 turquoise 1407
1 blue 201
5 brown 71
4 yellow 64
6 green 58
8 red 50
7 black 33
9 pink 21
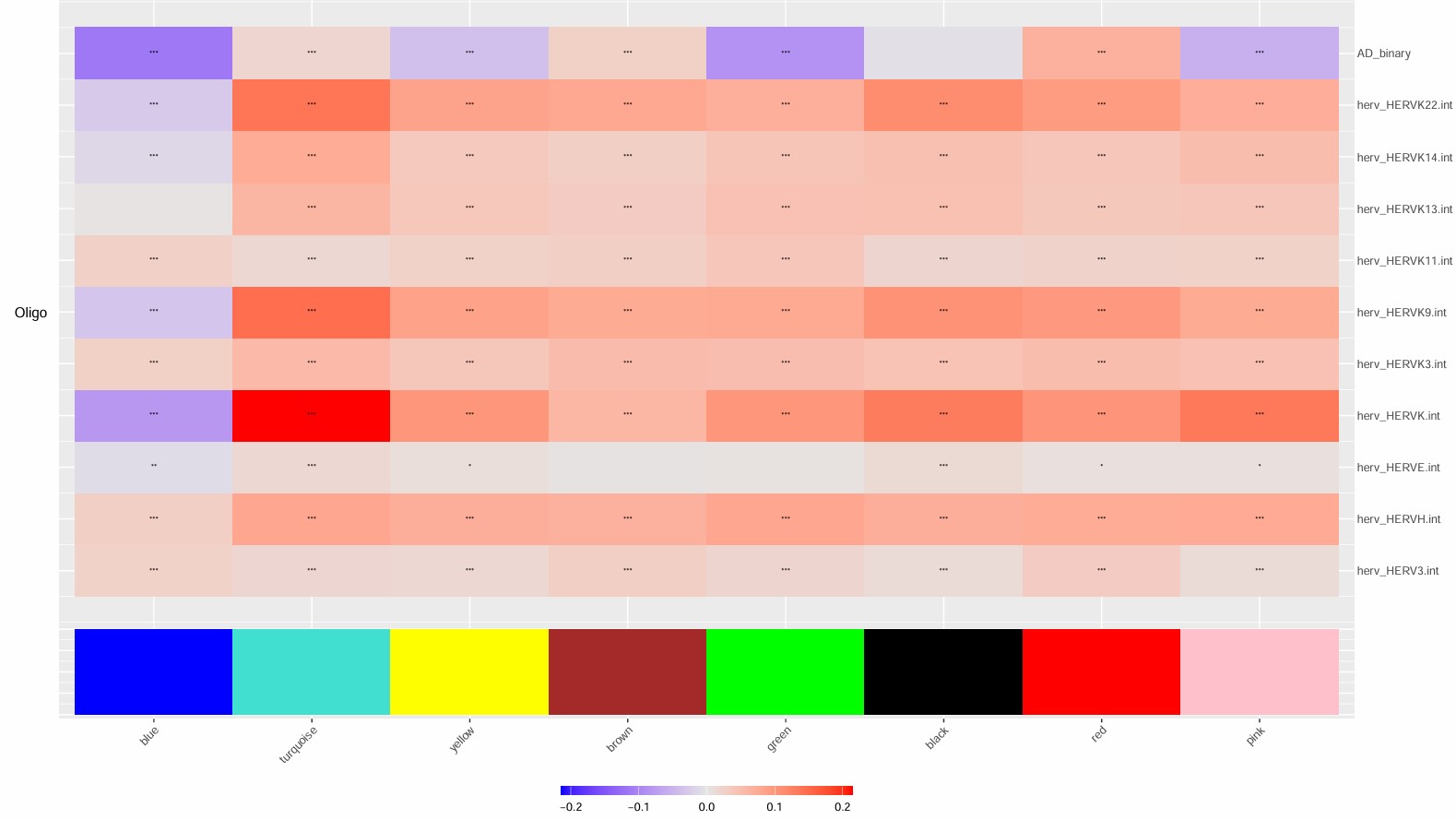
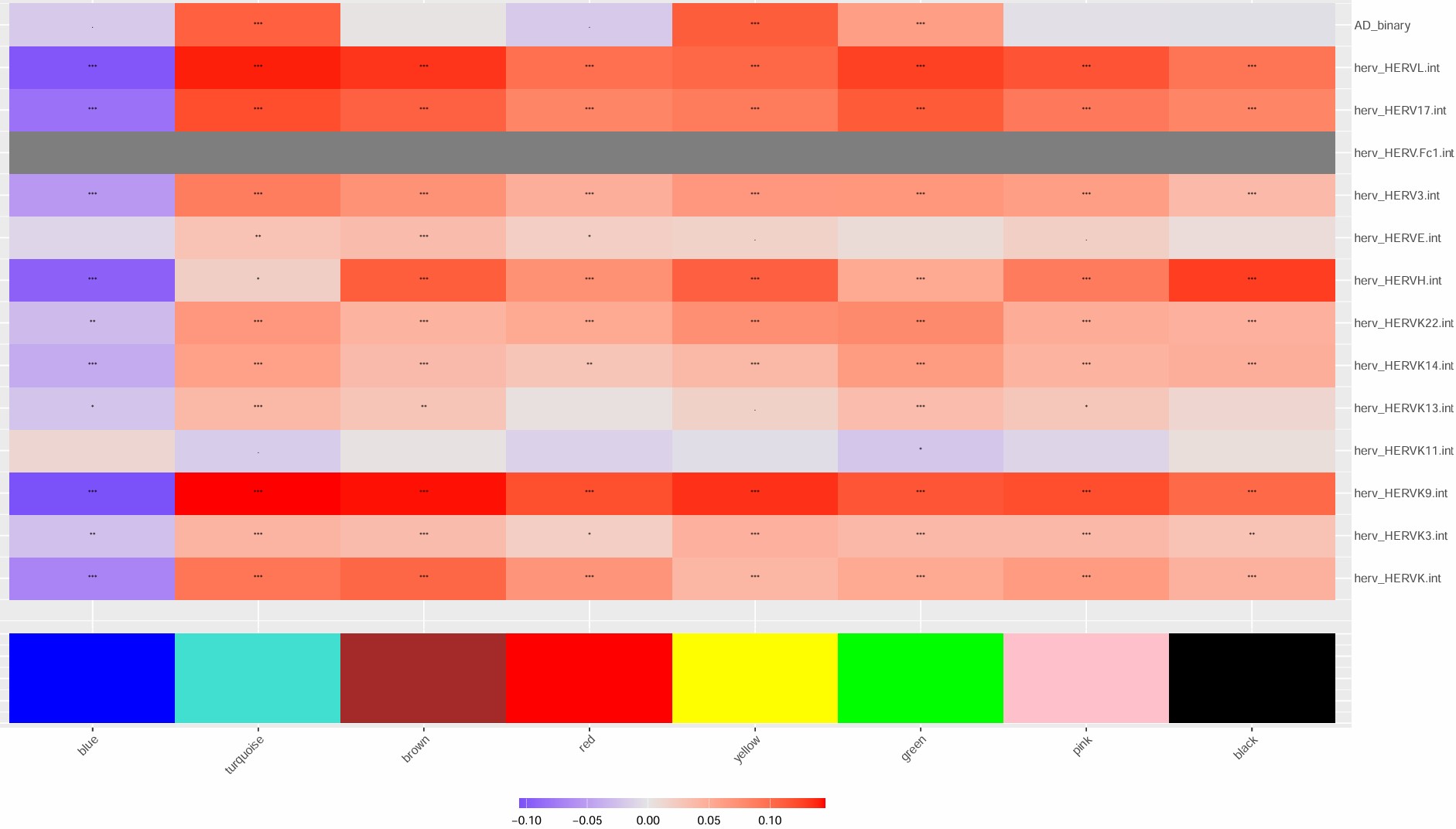
target_modules <- c("turquoise", "blue", "brown", "yellow", "green", "red")
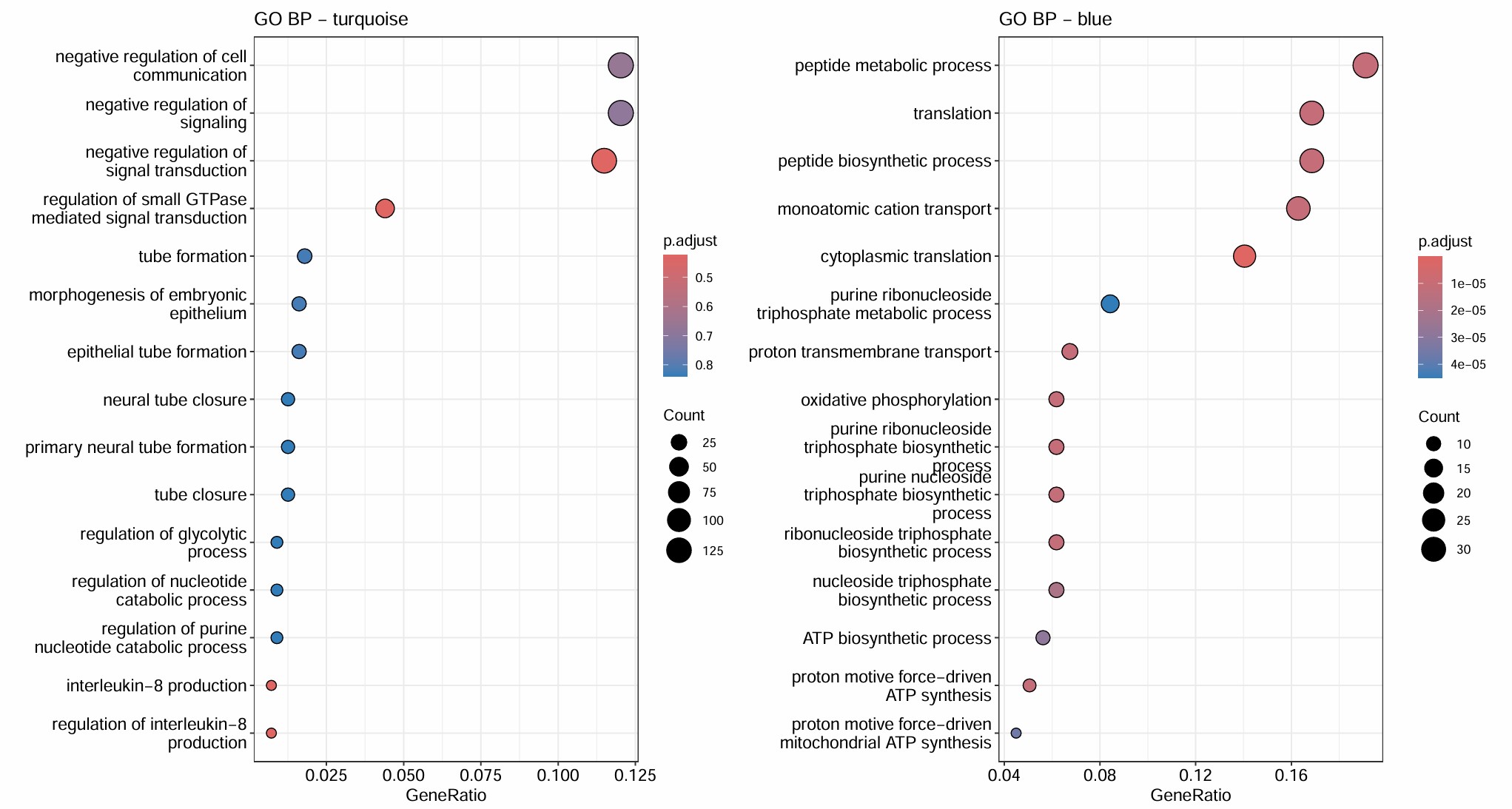
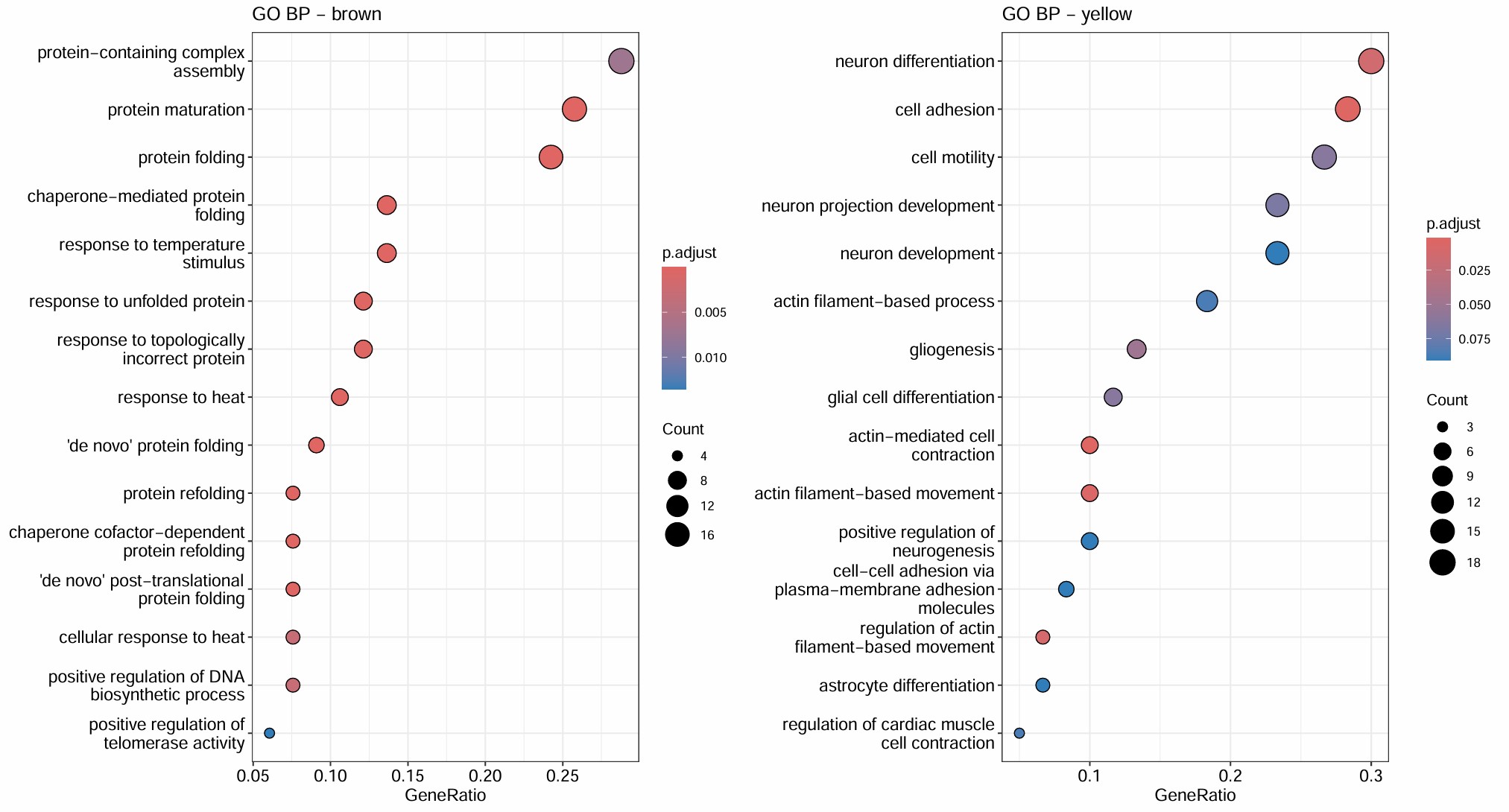
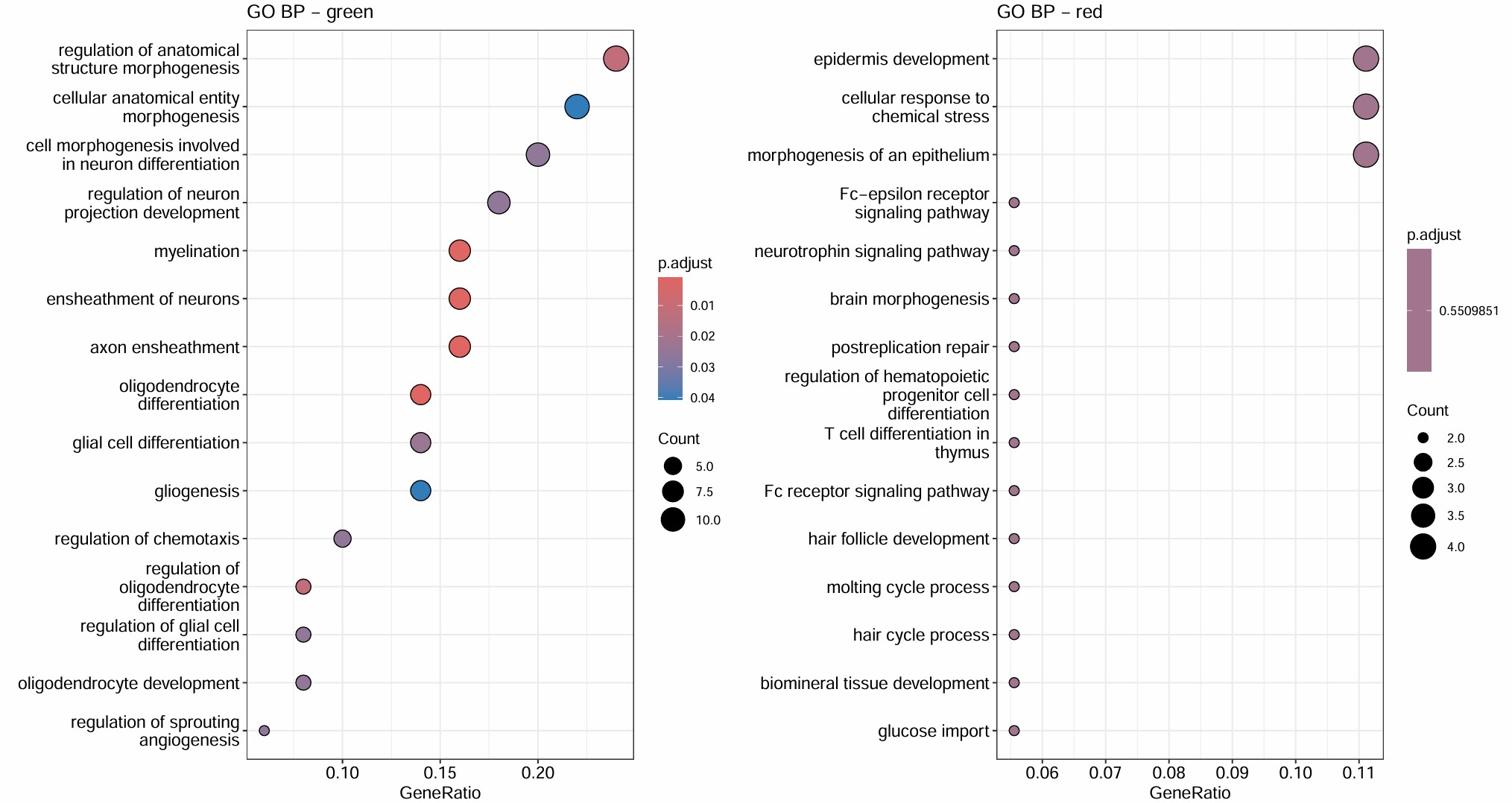
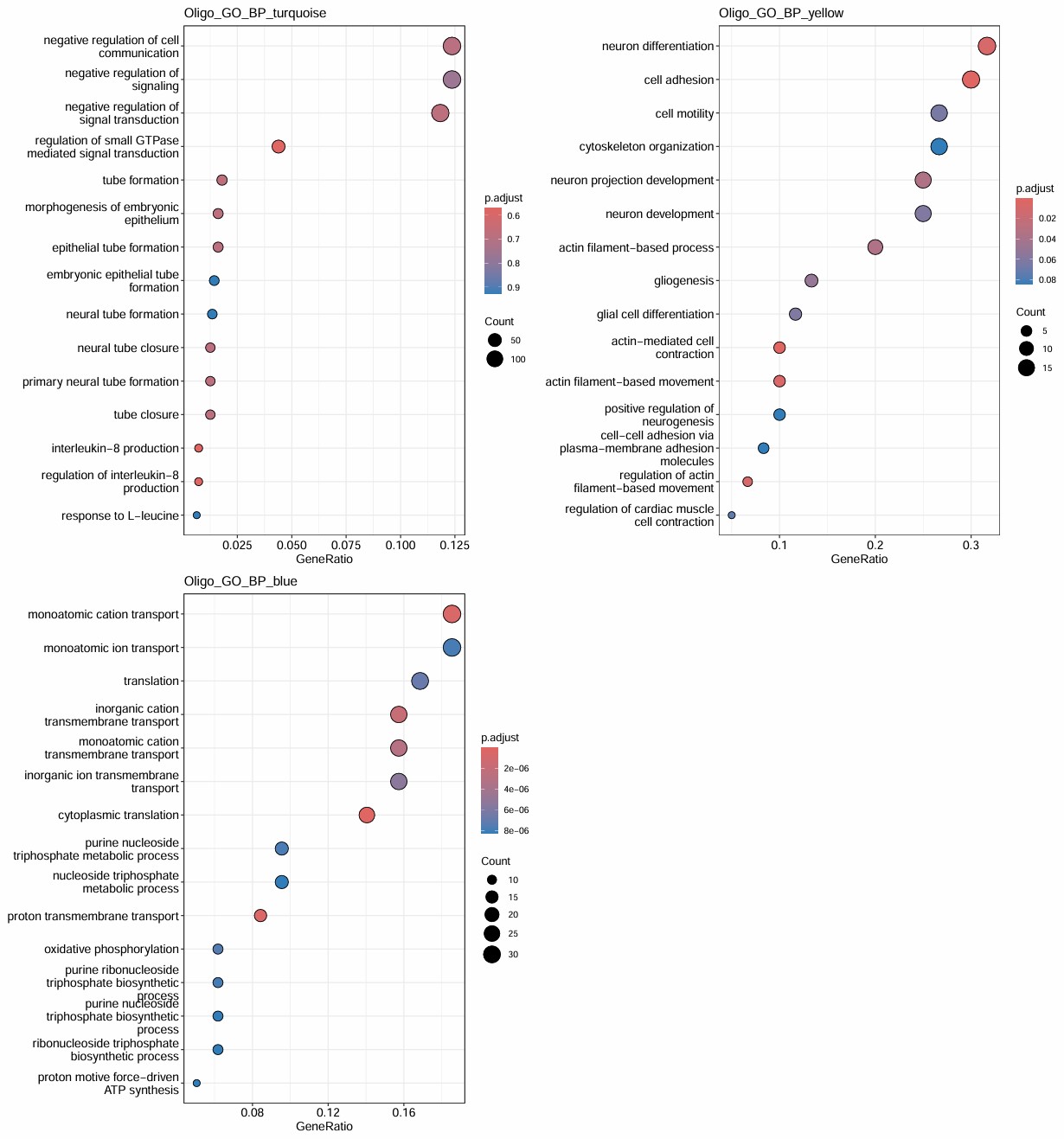
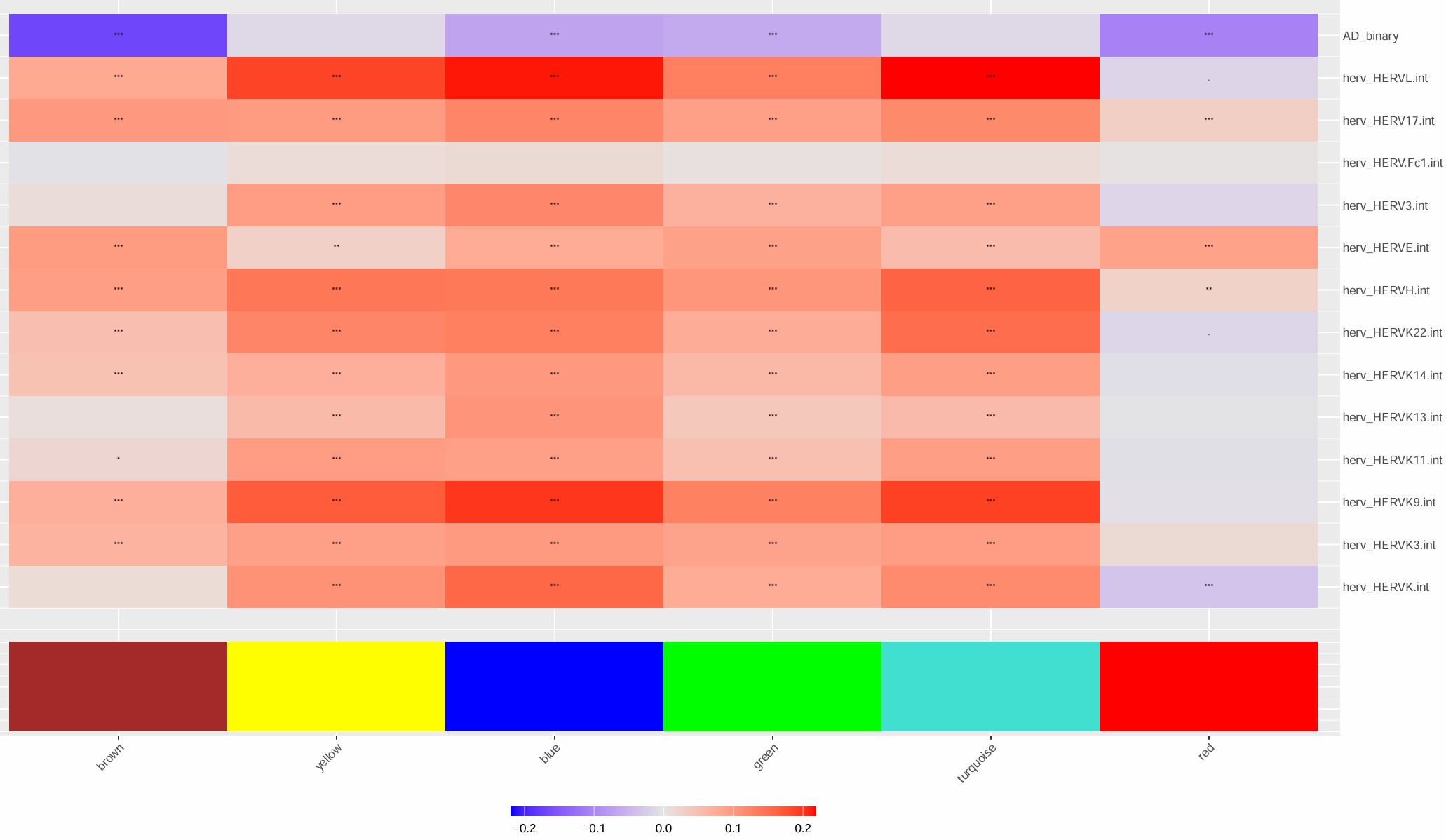
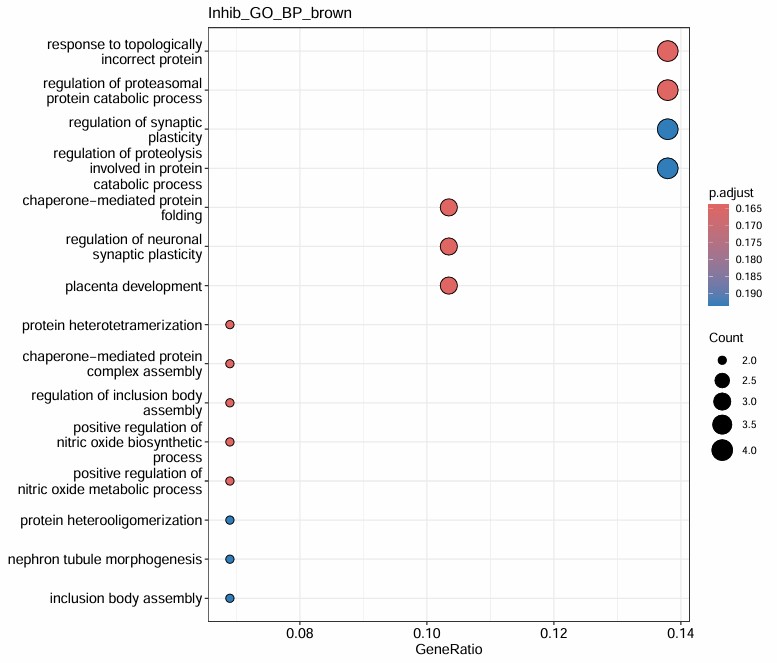
- turquoise和几乎所有hERV trait都是正相关,但基因数仍比较大(1400多个基因),GO结果里也有一些像small GTPase signaling/signal transduction negative regulation/actin cytoskeleton organization/IL-8 production这类看起来可能和AD有关的条目,但padj基本都不显著,属于广义状态程序
- blue的GO较集中且显著,都是关于能量代谢/蛋白合成的,hub genes也有不少核糖体/代谢相关基因:当hERV较高或AD状态更明显时,这套“翻译-氧化磷酸化-ATP合成”程序会下降
- green是少突分化/髓鞘形成模块,yellow是细胞黏附/神经突起/运动与分化模块,和hERVK都是正相关,和AD略偏负相关
- brown是蛋白折叠/热休克/蛋白稳态模块,和AD/hERV都是轻度正相关,更像是一个病理相关的模块
亚群层面分析:
- 对于hERV高表达的亚群,分为AD富集/不富集两种,如果该亚群hERV高表达且AD多,就对应brown/turquoise/blue这类AD和hERV同步变化的基因模块(蛋白稳态/广义hERV激活状态/稳态代谢受损),如果AD少就更像green/yellow这种不同步变化的(少突成熟/髓鞘化/分化或形态重塑)
- 理想情况是上面说的这样的,不过在我的亚群聚类时,至少在oligo的亚群里面,并没有出现某个亚群的AD特别富集,所以还是以模块表达分数为准
- 先看这个亚群的模块表达分数(就是上面得到的几个关键模块),看哪个模块在这个亚群里表达更高
- 把这个亚群和其它亚群做差异表达分析,得到一组亚群特异上调基因,看这些基因与各模块基因的重叠度,综合模块表达分数,确定这个亚群在哪个模块上特异表达
- 对DEG与目标模块内的基因取交集,再GO
load(file.path(res_dir, "oligo_WGCNA_try2.RData"))
seu_net <- seu
seu <- readRDS("/public/home/GENE_proc/wth/my_data/oligo.rds")
seu$subcluster <- seu$RNA_snn_res.0.5
wgcna_name <- "gene_net"
subcluster_col <- "subcluster"
target_clusters <- c("3", "5", "9")
modules_use <- c("brown", "turquoise", "blue", "green", "yellow")
# 取出模块基因
modules_df <- GetModules(seu_net, wgcna_name = wgcna_name)
gene_col <- intersect(c("gene_name", "gene", "feature"), colnames(modules_df))[1]
modules_df <- subset(modules_df, module %in% modules_use & module != "grey")
module_list <- split(modules_df[[gene_col]], modules_df$module)
module_list <- lapply(module_list, unique)
module_list <- lapply(module_list, intersect, rownames(seu[["RNA"]]))
# 给每个细胞打模块分数
for (m in modules_use) {
seu <- AddModuleScore(
seu,
features = list(module_list[[m]]),
assay = "RNA",
name = paste0(m, "_score_")
)
}
score_cols <- setNames(paste0(modules_use, "_score_1"), modules_use)
Idents(seu) <- seu[[subcluster_col]][, 1]
# 各亚群的模块平均分数
score_avg <- seu@meta.data[, c(subcluster_col, unname(score_cols)), drop = FALSE]
colnames(score_avg)[1] <- "subcluster"
score_avg <- score_avg %>%
group_by(subcluster) %>%
summarise(across(everything(), mean))
write.csv(score_avg, file.path(res_dir, "subcluster_module_scores.csv"), row.names = FALSE)
score_mat <- as.matrix(score_avg[, -1])
rownames(score_mat) <- score_avg$subcluster
colnames(score_mat) <- names(score_cols)
pdf(file.path(res_dir, "subcluster_module_scores.pdf"), width = 16, height = 16)
p <- pheatmap(
scale(score_mat),
cluster_rows = FALSE,
cluster_cols = FALSE,
main = "Scaled by module across subclusters"
)
print(p)
dev.off()
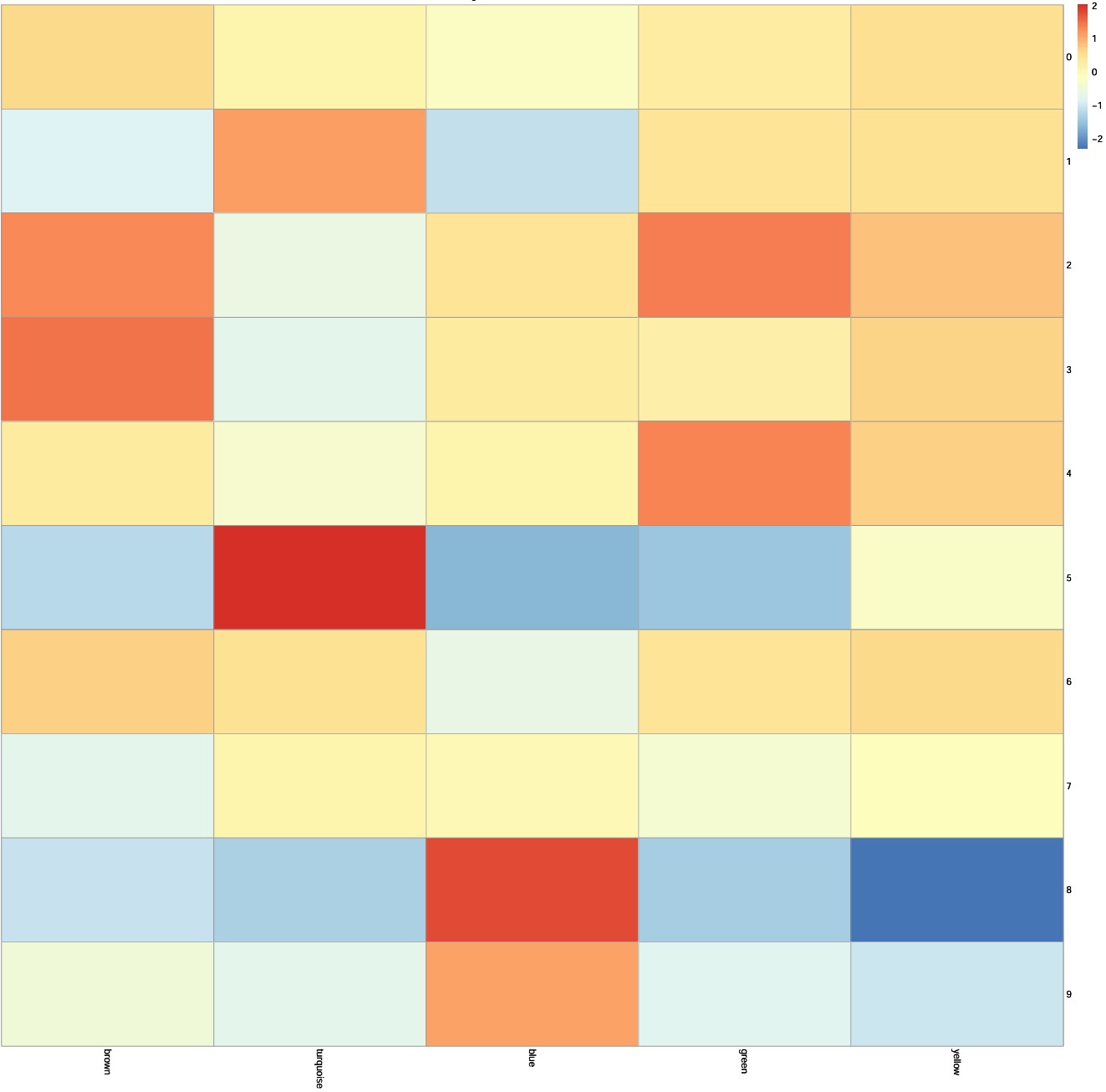
# 特异亚群与其它亚群的DEG
universe_genes <- unique(unlist(module_list))
Idents(seu) <- seu$subcluster
for (cl in target_clusters) {
other_ids <- setdiff(levels(Idents(seu)), cl)
deg <- FindMarkers(
seu,
ident.1 = cl,
ident.2 = other_ids,
assay = "RNA",
slot = "data",
test.use = "MAST",
# latent.vars = c("nCount_RNA", "percent_mito"),
min.pct = 0.1,
logfc.threshold = 0
)
deg$gene <- rownames(deg)
fc_col <- grep("^avg_log", colnames(deg), value = TRUE)[1]
deg_up <- subset(deg, p_val_adj < 0.05 & deg[[fc_col]] > 0.1)
write.csv(deg, file.path(res_dir, paste0("DEG_subcluster_", cl, "_all.csv")), row.names = FALSE)
write.csv(deg_up, file.path(res_dir, paste0("DEG_subcluster_", cl, "_up.csv")), row.names = FALSE)
target_mean <- colMeans(
seu@meta.data[seu[[subcluster_col]][, 1] == cl, unname(score_cols), drop = FALSE]
)
other_mean <- colMeans(
seu@meta.data[seu[[subcluster_col]][, 1] != cl, unname(score_cols), drop = FALSE]
)
score_tbl <- data.frame(
module = names(score_cols),
target_mean = unname(target_mean[unname(score_cols)]),
other_mean = unname(other_mean[unname(score_cols)]),
stringsAsFactors = FALSE
)
score_tbl$score_diff <- score_tbl$target_mean - score_tbl$other_mean
up_genes <- deg_up$gene
overlap_tbl <- data.frame(
module = names(score_cols),
overlap_n = 0,
gene_ratio = 0,
fisher_p = 1,
overlap_genes = "",
stringsAsFactors = FALSE
)
for (m in names(score_cols)) {
mod_genes <- module_list[[m]]
hit_genes <- intersect(up_genes, mod_genes)
a <- length(hit_genes)
b <- length(setdiff(up_genes, mod_genes))
c <- length(setdiff(mod_genes, up_genes))
d <- length(setdiff(universe_genes, union(up_genes, mod_genes)))
overlap_tbl[overlap_tbl$module == m, "overlap_n"] <- a
overlap_tbl[overlap_tbl$module == m, "gene_ratio"] <- ifelse(length(up_genes) > 0, a / length(up_genes), 0)
overlap_tbl[overlap_tbl$module == m, "overlap_genes"] <- paste(hit_genes, collapse = ";")
overlap_tbl[overlap_tbl$module == m, "fisher_p"] <- fisher.test(
matrix(c(a, b, c, d), nrow = 2),
alternative = "greater"
)$p.value
}
res_tbl <- left_join(score_tbl, overlap_tbl, by = "module") %>%
arrange(desc(score_diff), fisher_p)
write.csv(res_tbl, file.path(res_dir, paste0("subcluster_", cl, "_module_integration.csv")), row.names = FALSE)
}
save(module_list, universe_genes, file = file.path(res_dir, "0401.RData"))
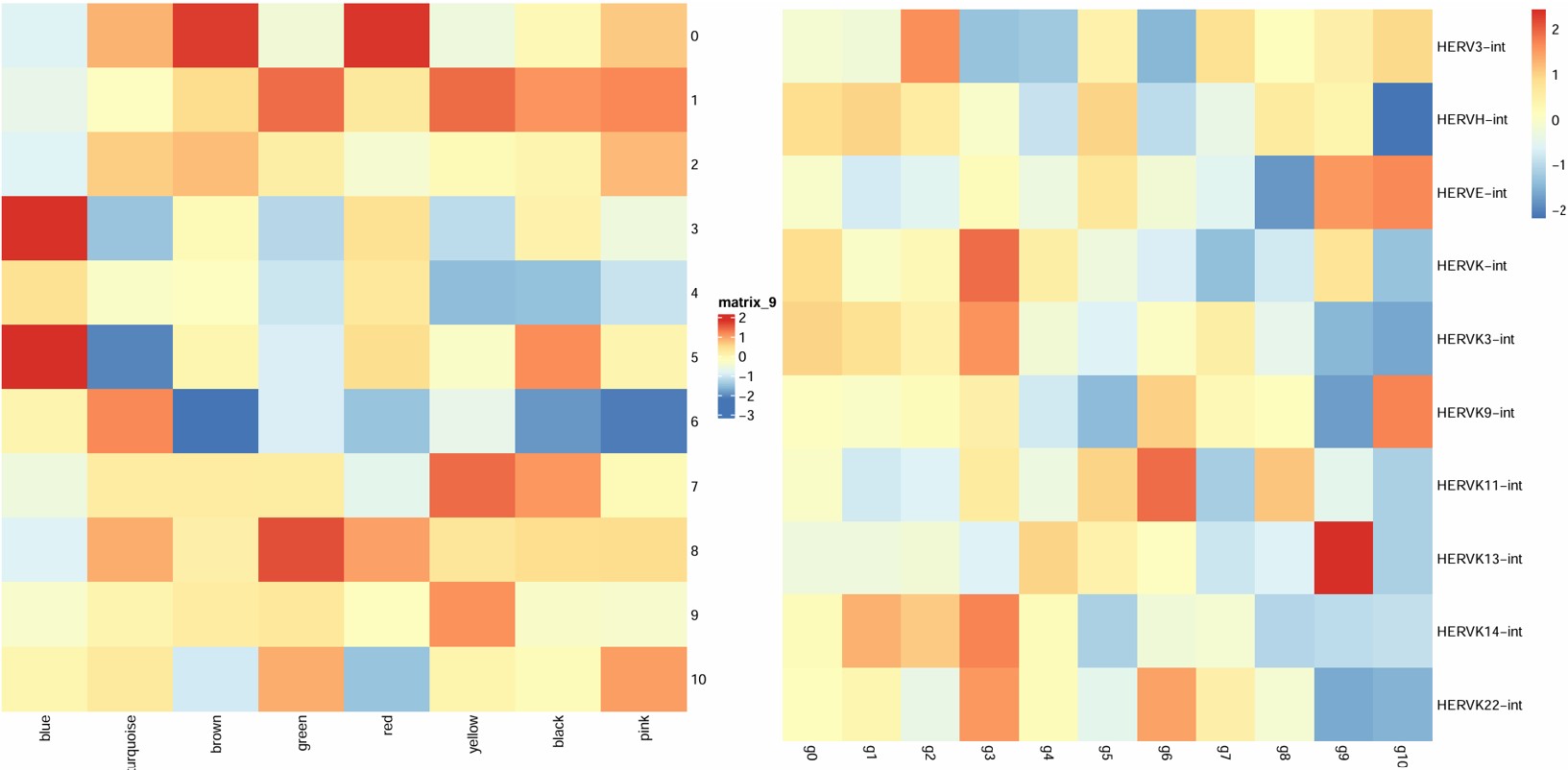
之前做的oligo各cluster的hERV平均表达量热图:
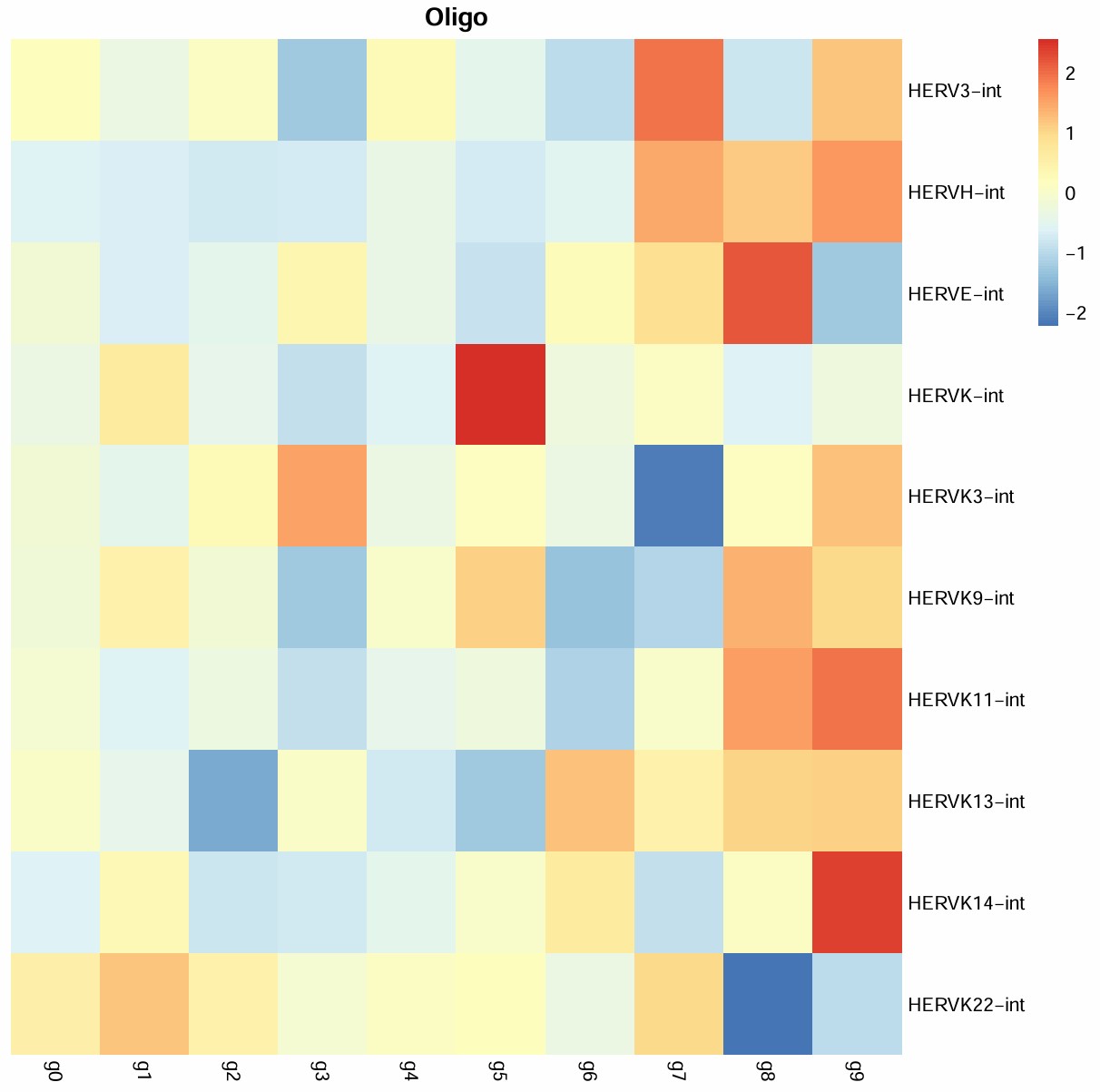
cluster 5:
- 从表达量热图中可以看到它的hERVK/hERVK9比较高
turquoise score_diff比较高,且上调DEG与turquoise的重叠较多- 最像广义hERV激活状态
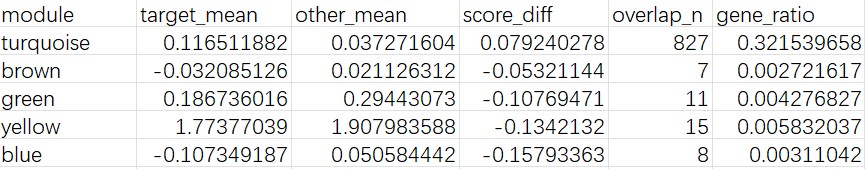
cluster 3:
- HERVK3比较高
- 上调基因包括ARHGAP24/CTNNA2/PTPRM/DCC/BCAN/ITPR2/PLEKHH2,属于细胞黏附/细胞骨架/发育的模块,与yellow模块的GO结果吻合
- 像结构重塑/形态变化/分化相关亚群
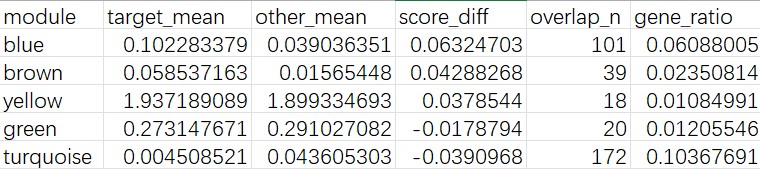
cluster 9:
- HERVK家族普遍较高
- 虽然上调基因有一些偏向神经元的标记基因,不过画了一下dotplot发现整体上更偏向oligo而不是神经元
- blue模块分数较高
target_module_map <- c(
"5" = "turquoise",
"3" = "yellow",
"9" = "blue"
)
plot_list <- list()
for (cl in names(target_module_map)) {
deg_up <- read.csv(
paste0("C:\\Users\\17185\\Desktop\\fsdownload\\DEG_subcluster_", cl, "_up.csv"),
stringsAsFactors = FALSE
)
target_module <- target_module_map[[cl]]
gene_use <- intersect(deg_up$gene, module_list[[target_module]])
write.csv(
data.frame(gene = gene_use),
paste0("C:\\Users\\17185\\Desktop\\fsdownload\\subcluster_", cl, "_", target_module, "_intersect_genes.csv"),
row.names = FALSE
)
ego <- enrichGO(
gene = gene_use,
universe = universe_genes,
OrgDb = org.Hs.eg.db,
keyType = "SYMBOL",
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 1,
qvalueCutoff = 1,
readable = TRUE
)
ego_df <- as.data.frame(ego)
write.csv(
ego_df,
paste0("C:\\Users\\17185\\Desktop\\fsdownload\\subcluster_", cl, "_", target_module, "_GO_BP.csv"),
row.names = FALSE
)
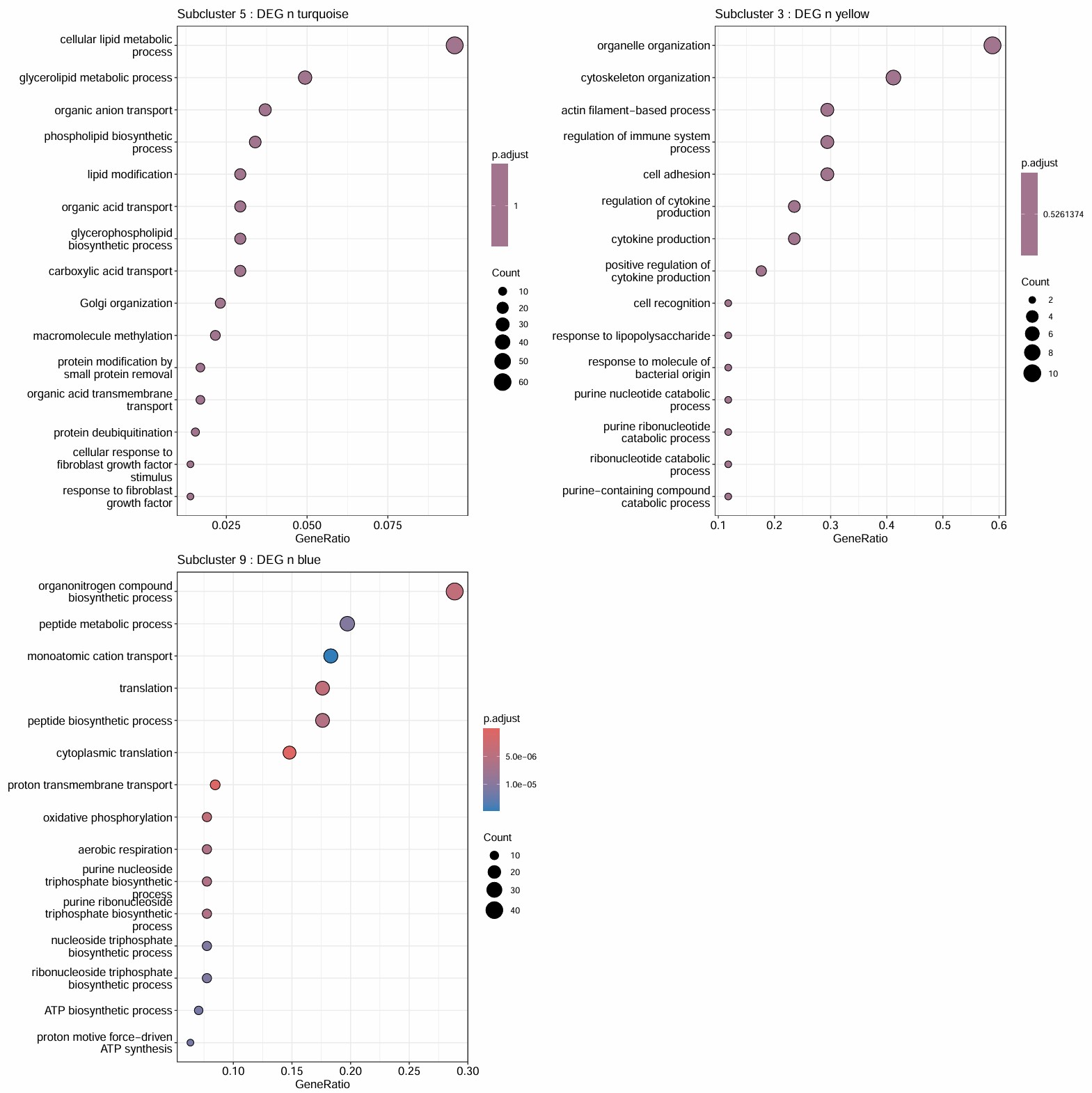
plot_list[[cl]] <- dotplot(ego, showCategory = 15) +
ggtitle(paste0("Subcluster ", cl, " : DEG ∩ ", target_module))
}
pdf(file.path(res_dir, "oligo_359_GO_summary.pdf"), width = 16, height = 16)
p <- wrap_plots(plot_list, ncol = 2)
print(p)
dev.off()
-
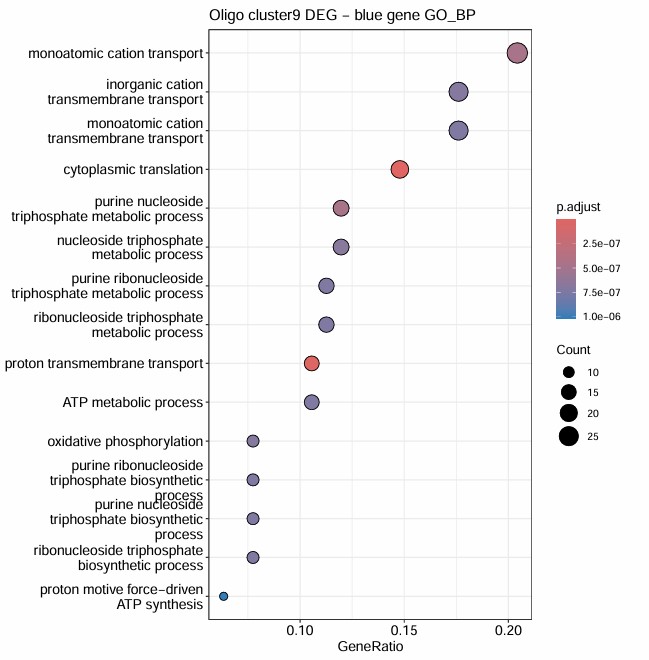
cluster 9的GO结果是唯一真正显著的,且GO富集条目与blue亚群吻合,说明它最可能代表的是:一个翻译活跃、线粒体呼吸活跃、能量代谢强、同时又保留成熟髓鞘marker的少突亚群
结合前面的hERV表达量热图,可以理解成:cluster 9虽然也是hERV-high,但它对应的不是 “HERVK-int驱动的广义激活状态”,而更像一种“成熟髓鞘化背景下的hERV表达模式”
-
cluster 5的GO结果虽然不显著,但也符合turquoise模块的GO结果,在功能上呈现出脂质代谢、膜转运、去泛素化和FGF反应等倾向。说明交集基因虽然多,但功能比较分散
它是最像hERV激活型的亚群,只是它的功能解释更适合写成脂质与膜代谢重塑、广义信号调节
-
cluster 3的GO结果与5类似,它确实更偏向细胞骨架/黏附/actin重塑/与外界刺激或炎症反应有关的调节,像是一个结构重塑/黏附变化/分化过程中的亚群
总结:hERV高表达在少突细胞内并不对应单一病理程序,而是和不同的细胞状态相联系
-
cluster 5:hERVK/K9主导的广义激活型
功能倾向:脂质代谢、膜转运、信号/修饰重塑
-
cluster 3:HERVK3相关的结构重塑型
功能倾向:actin、黏附、形态/细胞识别
-
cluster 9:多种hERV相关的成熟髓鞘化/高代谢型
功能:翻译、氧化磷酸化、ATP合成
方案2
mixed_herv_features <- c("HERV3-int", "HERVH-int", "HERVE-int", "HERVK-int", "HERVK3-int", "HERVK9-int", "HERVK11-int", "HERVK13-int", "HERVK14-int", "HERVK22-int")
# 过滤掉表达比例<5%的基因(为防止把加进去的hERV也过滤掉,所以就不在SetupForWGCNA时过滤)
rna_counts <- LayerData(seu, assay = "RNA", layer = "counts")
feature_rowsum <- Matrix::rowSums(rna_counts > 0)
retained_f_low <- feature_rowsum > ncol(seu) * 0.05
genes_keep <- names(feature_rowsum)[retained_f_low]
seu[["RNA"]] <- subset(seu[["RNA"]], features = genes_keep)
# 构建混合assay:RNA gene + hERV feature
rna_counts <- LayerData(seu, assay = "RNA", layer = "counts")
rna_data <- LayerData(seu, assay = "RNA", layer = "data")
herv_counts <- LayerData(seu, assay = herv_assay, layer = "counts")[mixed_herv_features, , drop = FALSE]
herv_data <- LayerData(seu, assay = herv_assay, layer = "data")[mixed_herv_features, , drop = FALSE]
rownames(herv_counts) <- paste0("HERV__", rownames(herv_counts))
rownames(herv_data) <- paste0("HERV__", rownames(herv_data))
mixed_counts <- rbind(rna_counts, herv_counts)
mixed_data <- rbind(rna_data, herv_data)
seu[["RNA_HERV"]] <- CreateAssayObject(counts = mixed_counts)
LayerData(seu, assay = "RNA_HERV", layer = "data") <- mixed_data
DefaultAssay(seu) <- "RNA_HERV"
mixed_features <- rownames(seu[["RNA_HERV"]])
# 重复前述步骤
seu <- SetupForWGCNA(
seu,
gene_select = "custom",
gene_list = mixed_features,
assay = "RNA_HERV",
wgcna_name = "mixed_net"
)
seu <- MetacellsByGroups(
seurat_obj = seu,
group.by = c("celltype", "orig.ident"),
ident.group = "celltype",
assay = "RNA_HERV",
layer = "data",
reduction = "pca",
dims = 1:30,
k = 25,
max_shared = 10,
min_cells = 50,
mode = "average",
wgcna_name = "mixed_net"
)
seu <- NormalizeMetacells(seu, wgcna_name = "mixed_net")
seu <- SetDatExpr(
seu,
group_name = celltype_name,
group.by = "celltype",
use_metacells = TRUE,
assay = "RNA_HERV",
layer = "data",
wgcna_name = "mixed_net"
)
grep("^HERV__", rownames(seu))
seu <- TestSoftPowers(
seu,
networkType = "signed",
wgcna_name = "mixed_net"
)
power_table_mixed <- GetPowerTable(seu)
soft_power_mixed <- power_table_mixed$Power[which(power_table_mixed$SFT.R.sq >= 0.8)[1]]
if (is.na(soft_power_mixed)) {
soft_power_mixed <- power_table_mixed$Power[which.max(power_table_mixed$SFT.R.sq)]
}
plot_list <- PlotSoftPowers(seu)
pdf(file.path(res_dir, "oligo_soft_power2.pdf"), width = 16, height = 16)
p <- patchwork::wrap_plots(plot_list, ncol = 2)
print(p)
dev.off()
save(seu, file = file.path(res_dir, "oligo_WGCNA2_before_ConstructNetwork.RData"))
seu <- ConstructNetwork(
seu,
assay = "RNA_HERV",
soft_power = soft_power_mixed,
networkType = "signed",
tom_name = "seu_TOM",
overwrite_tom = TRUE,
wgcna_name = "mixed_net"
)
seu <- ScaleData(
seu,
assay = "RNA_HERV",
features = rownames(seu[["RNA_HERV"]]),
)
seu <- ModuleEigengenes(
seu,
# group.by.vars = "orig.ident",
assay = "RNA_HERV",
wgcna_name = "mixed_net"
)
seu <- ModuleConnectivity(
seu,
group.by = "celltype",
group_name = celltype_name,
assay = "RNA_HERV",
wgcna_name = "mixed_net"
)
save(seu, file = file.path(res_dir, "oligo_WGCNA2.RData"))
module_genes_df <- GetModules(seu)
hub_genes_df <- GetHubGenes(seu, n_hubs = 50)
modules <- unique(module_genes_df$module)
genes_list <- list()
for(single_module in modules){
module_genes <- module_genes_df %>%
filter(module == single_module) %>%
pull(gene_name) %>%
unique()
genes_list[[single_module]]$module_genes <- module_genes
hub_genes <- hub_genes_df %>%
filter(module == single_module) %>%
pull(gene_name) %>%
unique()
genes_list[[single_module]]$hub_genes <- hub_genes
}
TOM <- GetTOM(seu)
target_herv <- grepl("^HERV--", colnames(TOM)) # 目标hERV
target_gene <- !grepl("^HERV--", rownames(TOM)) # gene
universe_genes <- rownames(TOM)[target_gene]
herv_neighbors <- as.data.frame(TOM[target_gene, target_herv]) %>%
rownames_to_column("rowname") %>%
as_tibble() %>%
column_to_rownames("rowname")
res_list <- list()
for(herv in colnames(herv_neighbors)){
herv_gene <- herv_neighbors[, herv, drop = FALSE]
colnames(herv_gene) <- "value"
herv_module <- subset(module_genes_df, gene_name == herv)$module
print(paste0(herv, "所属的模块为:", herv_module))
module_genes <- subset(module_genes_df, module == herv_module)$gene_name
res_list[[herv]]$module <- herv_module
cor_gene <- subset(herv_gene, rownames(herv_gene) %in% module_genes) %>%
filter(abs(value) > 0.02) %>%
arrange(desc(value)) %>%
head(n = 300) %>%
rownames_to_column("gene")
res_list[[herv]]$cor <- cor_gene
res_list[[herv]]$cor_range <- c(max(cor_gene$value), min(cor_gene$value))
res_list[[herv]]$gene <- cor_gene$gene
}
save(genes_list, res_list, universe_genes, file = file.path(res_dir, "oligo_WGCNA2_res.RData"))
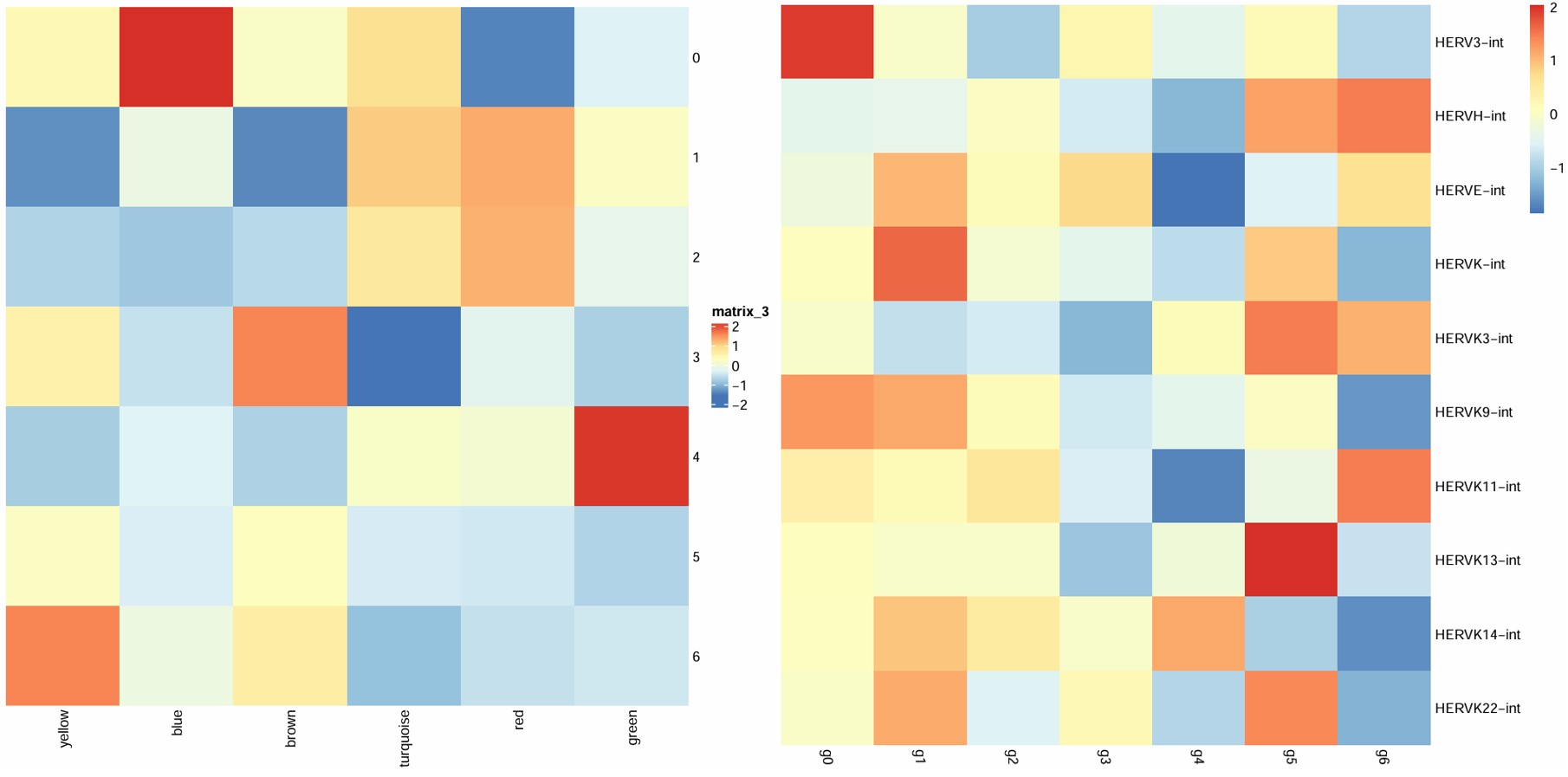
"HERV--HERV3-int所属的模块为:grey"
"HERV--HERVH-int所属的模块为:blue"
"HERV--HERVE-int所属的模块为:grey"
"HERV--HERVK-int所属的模块为:turquoise"
"HERV--HERVK3-int所属的模块为:grey"
"HERV--HERVK9-int所属的模块为:turquoise"
"HERV--HERVK11-int所属的模块为:blue"
"HERV--HERVK13-int所属的模块为:grey"
"HERV--HERVK14-int所属的模块为:grey"
"HERV--HERVK22-int所属的模块为:turquoise"
GO富集分析:
- 对HERVK/HERVK9/HERVK22/HERVH/HERVK11这几个不在grey模块(grey模块就是指比较模糊/没法聚类的基因)里的hERV相关性前300的基因做GO
- 对它们所在的模块做GO
# 前300基因
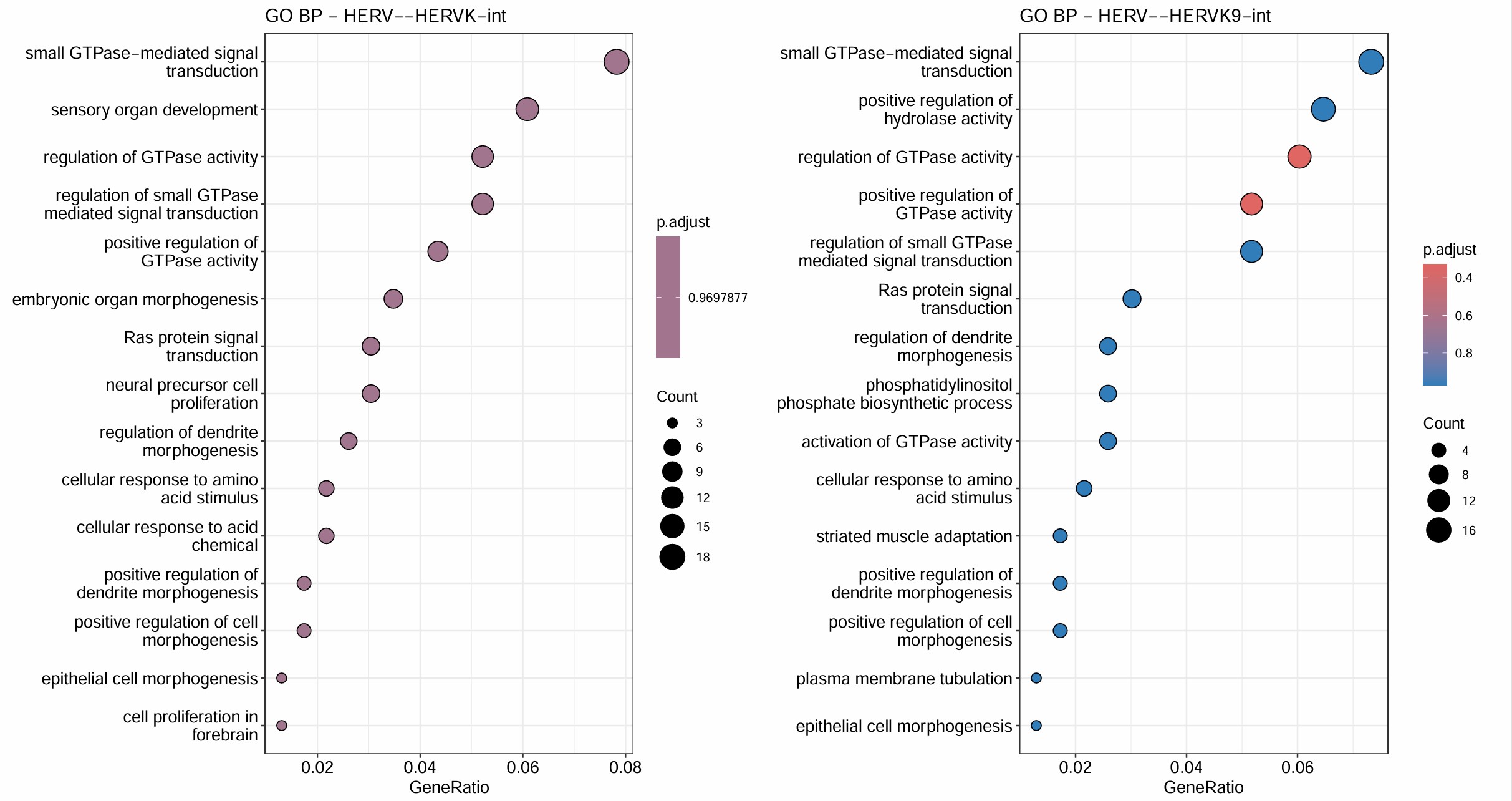
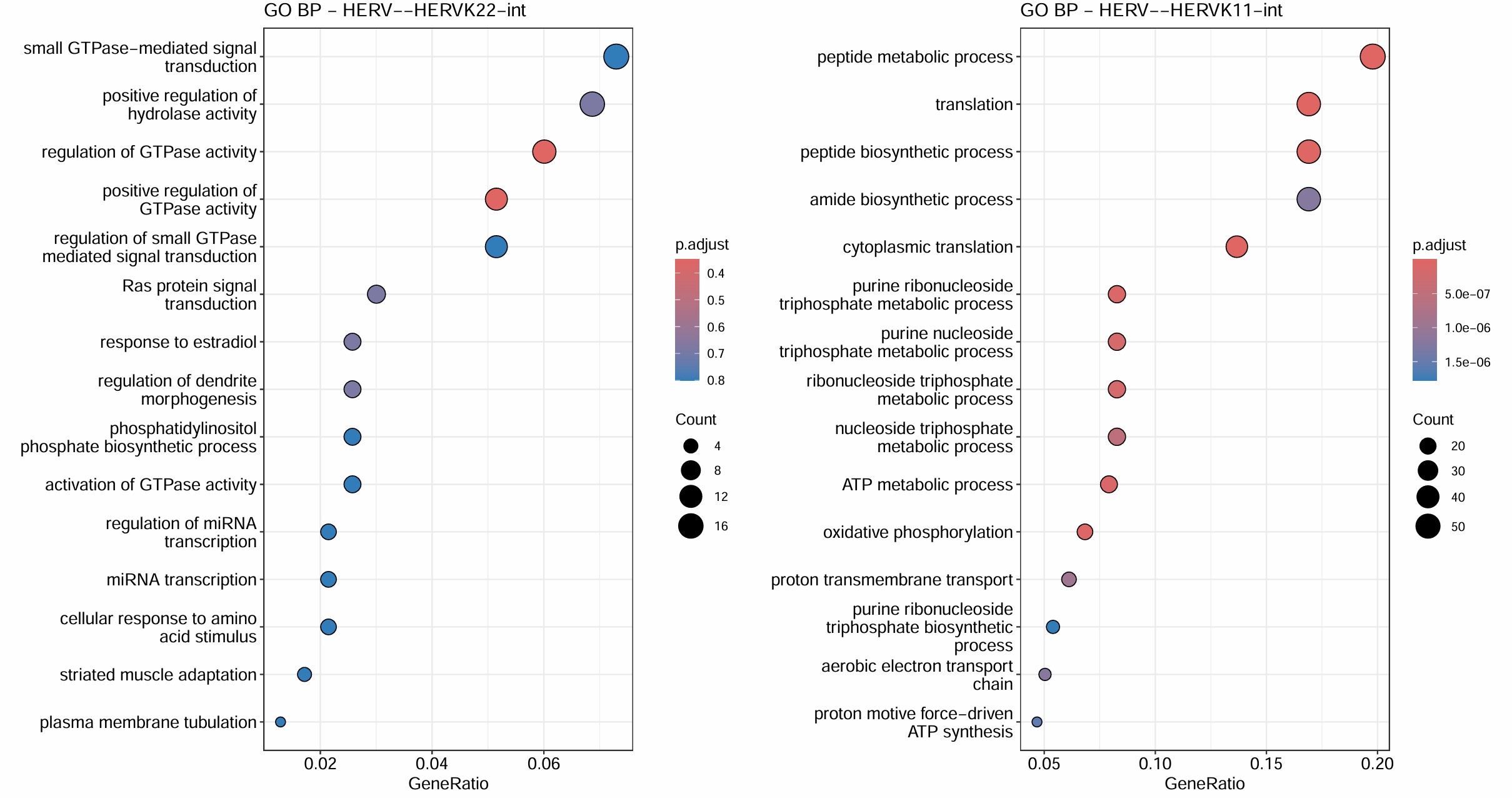
target_hervs <- c("HERV--HERVK-int", "HERV--HERVK9-int", "HERV--HERVK22-int", "HERV--HERVK11-int", "HERV--HERVH-int")
go_list <- lapply(target_hervs, function(m) {
run_go_one(
gene_vec = res_list[[m]]$gene,
universe_genes = universe_genes,
ont = "BP"
)
})
names(go_list) <- target_hervs
res <- list()
for (m in target_hervs) {
ego <- go_list[[m]]
if (is.null(ego)) next
res[[m]] <- dotplot(ego, showCategory = 15) +
ggtitle(paste0("GO BP - ", m))
}
pdf(file.path(res_dir, "oligo_hERV_gene_summary.pdf"), width = 15, height = 24)
p <- wrap_plots(res, ncol = 2)
print(p)
dev.off()
oligo_hERV_gene_summary <- lapply(target_hervs, function(m) {
ego <- go_list[[m]]
if (is.null(ego)) return(NULL)
go_df <- as.data.frame(ego)
go_df <- go_df[order(go_df$p.adjust), ]
herv_list <- res_list[[m]]
data.frame(
herv = m,
module = as.character(herv_list$module),
cor_gene = paste(herv_list$gene, collapse = "; "),
max_cor = herv_list$cor_range[1],
min_cor = herv_list$cor_range[2],
GO = paste(go_df$Description[1:20], collapse = "; "),
GO_padj = paste(go_df$p.adjust[1:20], collapse = "; "),
stringsAsFactors = FALSE
)
}) %>% bind_rows()
write.csv(oligo_hERV_gene_summary, file.path(res_dir, "oligo_hERV_gene_summary.csv"), row.names = FALSE)
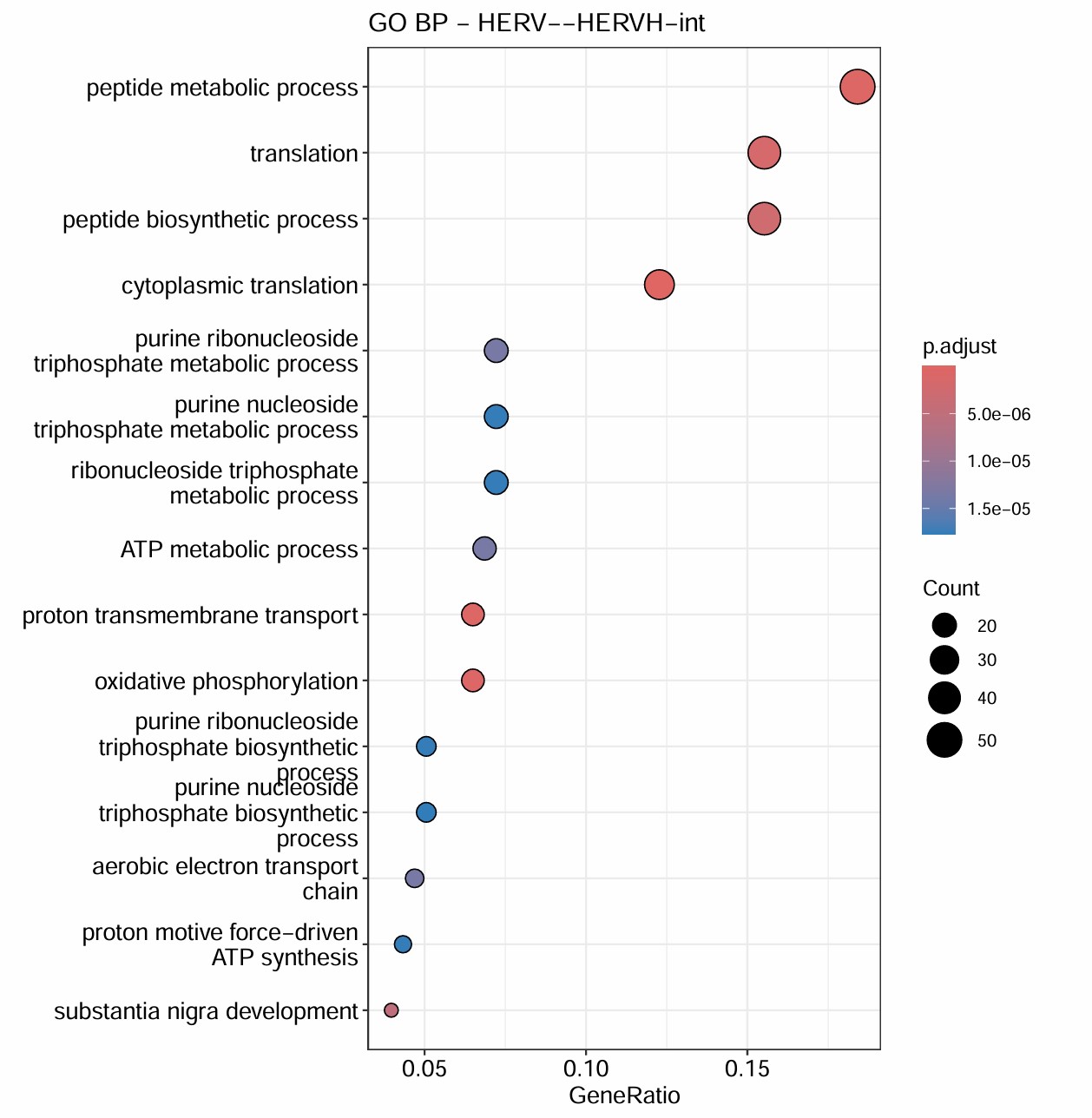
# 它们所在的模块
target_modules <- unique(as.character(oligo_hERV_gene_summary$module))
go_list <- lapply(target_modules, function(m) {
run_go_one(
gene_vec = genes_list[[m]]$module_genes,
universe_genes = universe_genes,
ont = "BP"
)
})
names(go_list) <- target_modules
res <- list()
for (m in target_modules) {
ego <- go_list[[m]]
if (is.null(ego)) next
res[[m]] <- dotplot(ego, showCategory = 15) +
ggtitle(paste0("GO BP - ", m))
}
pdf(file.path(res_dir, "oligo_hERV_module_summary.pdf"), width = 16, height = 8)
p <- wrap_plots(res, ncol = 2)
print(p)
dev.off()
oligo_hERV_module_summary <- lapply(target_modules, function(m) {
ego <- go_list[[m]]
if (is.null(ego)) return(NULL)
go_df <- as.data.frame(ego)
go_df <- go_df[order(go_df$p.adjust), ]
module_list <- genes_list[[m]]
data.frame(
module = m,
n_gene = length(module_list$module_genes),
hub_gene = paste(module_list$hub_genes, collapse = "; "),
GO = paste(go_df$Description[1:20], collapse = "; "),
GO_padj = paste(go_df$p.adjust[1:20], collapse = "; "),
stringsAsFactors = FALSE
)
}) %>% bind_rows()
write.csv(oligo_hERV_module_summary, file.path(res_dir, "oligo_hERV_module_summary.csv"), row.names = FALSE)
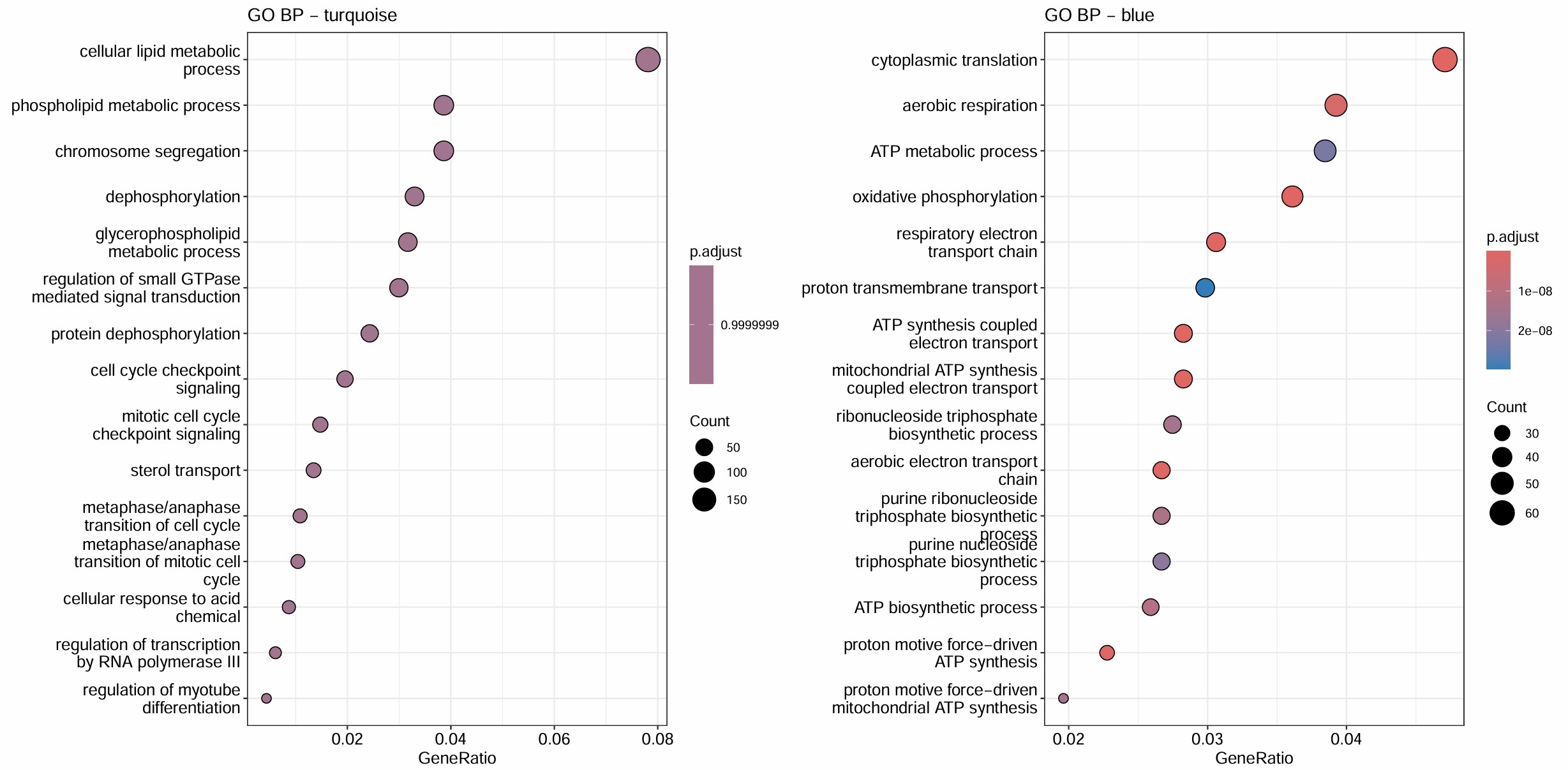
综合上面的结果,HERVH-int和HERVK11-int的GO结果最显著,且它们都在blue这个module,对该模块的GO分析结果也显著集中于蛋白翻译+线粒体能量代谢。另外几个都在turquoise模块,不过这个模块的GO结果与方案1的类似,都比较模糊
-
一个比较大的问题就是与hERV相关的前300个基因的相关性在0.05±0.02左右,这个相关性应如何衡量?
这个相关性实际上不是前面线性拟合时得到的相关性,而是TOM相似度,这种计算方法得到的数值会比普通相关系数小很多,因此这个值其实不能说是“弱相关”
接下来的GO分析与上面方案1的类似,都是计算目标亚群与其它亚群的DEG,然后看目标亚群的高表达hERV相邻的基因与DEG的重叠度,取两者重合的部分作GO
共表达网络
选hERV特异亚群和显著神经相关模块进行GO分析
以下结果,如果family和group的没有太大差别,就只展示family的结果,后期也以family为主进行分析
hERV表达量热图是按行看——每个hERV家族在各亚群中的相对表达水平,模块分数热图是按列看,每个模块在各亚群中的相对表达水平(其实只是行列倒过来了,意义相同)
Oligo
汇总一下之前的记录:
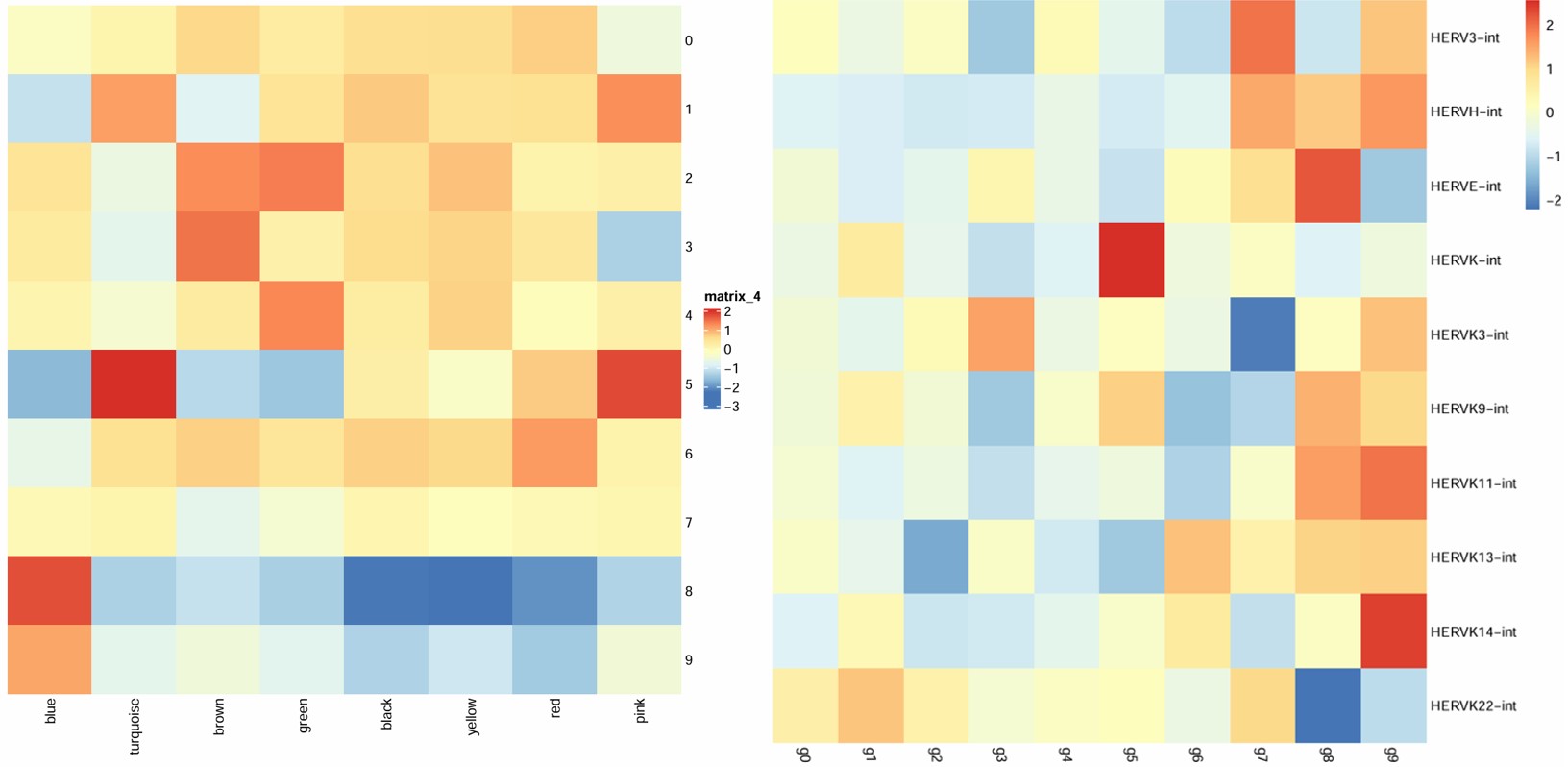
Astro
- 0在blue表达量高,blue和hERVK有正相关,和AD微弱负相关,可作为目标亚群
- 1在red表达量高,但red和hERV相关性较弱,不如0+blue
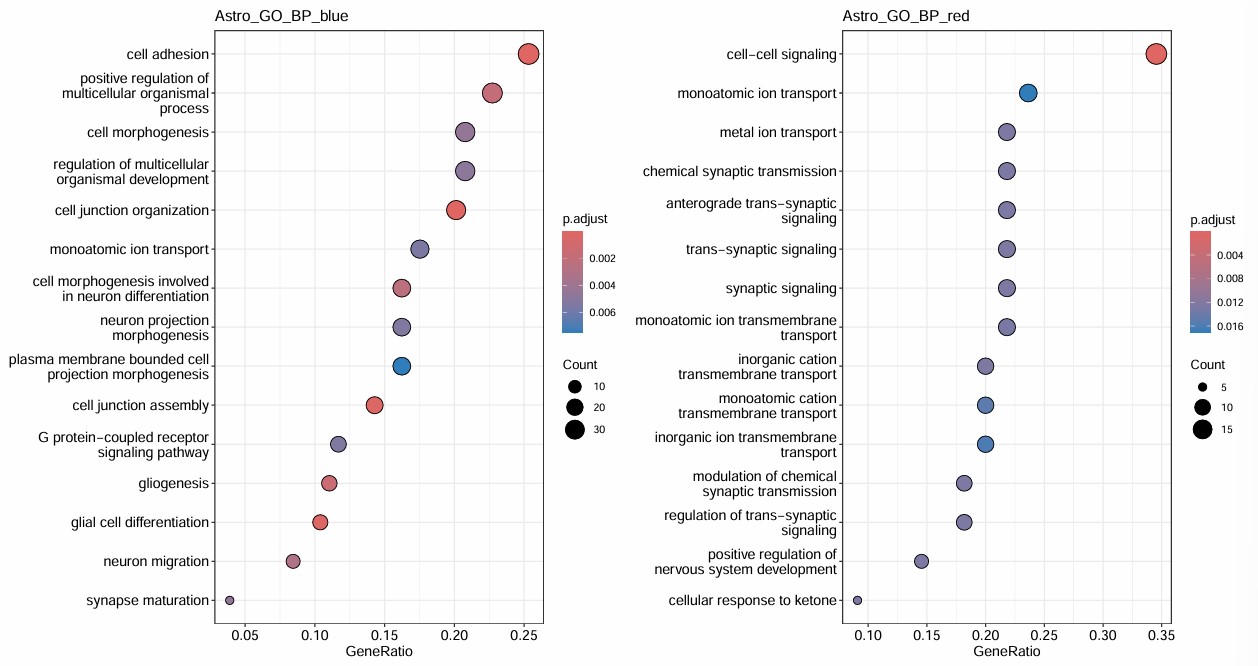
- blue和red都有神经相关的条目,其中blue神经相关条目看起来非常多
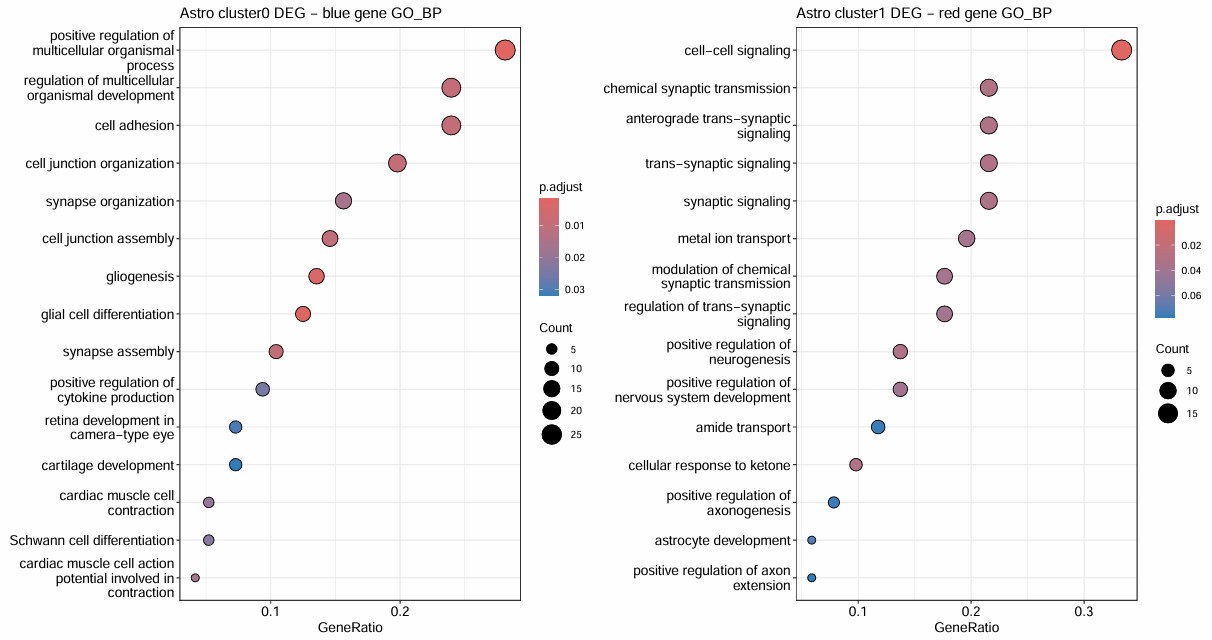
- cluster0有15000个细胞,占astro的3/4,AD占比很平均
- cluster1有3000多个细胞,AD稍微多一些(比astro整体AD占比多3%)
- 但是它们的GO聚类结果很好,blue和神经功能显著相关,red也较显著,和信号传导相关
OPC
- 6在turquoise表达量高,turquoise和hERVK有正相关,和AD无相关,可作为目标亚群
- 8没有特别高的,只有yellow有点点高,但yellow和hERV相关性不如6+turquoise
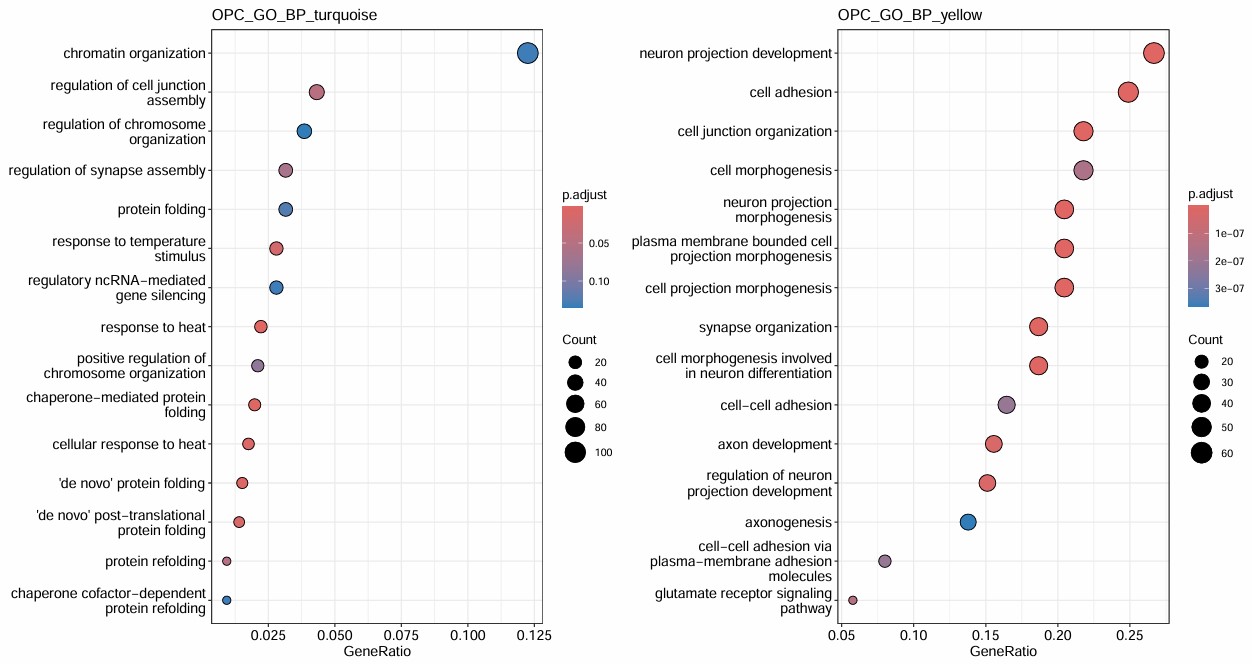
- yellow和astro的blue几乎一模一样,turquoise更偏普通功能,也有染色体结构/转录翻译这些
- 然而不知道为什么OPC的6/8亚群的DEG只有十几个,取交集后就只剩下个位数了
Excit
- 6hERV下调,在blue/green/magenta/pink模块也下调,在black/red/yellow模块上调
- blue/green/magenta/pink与hERV都正相关
- black与hERV负相关程度很大,red其次,yellow相关程度较低
- 因此可以看看6与blue/green/magenta/pink/black/red这几个模块
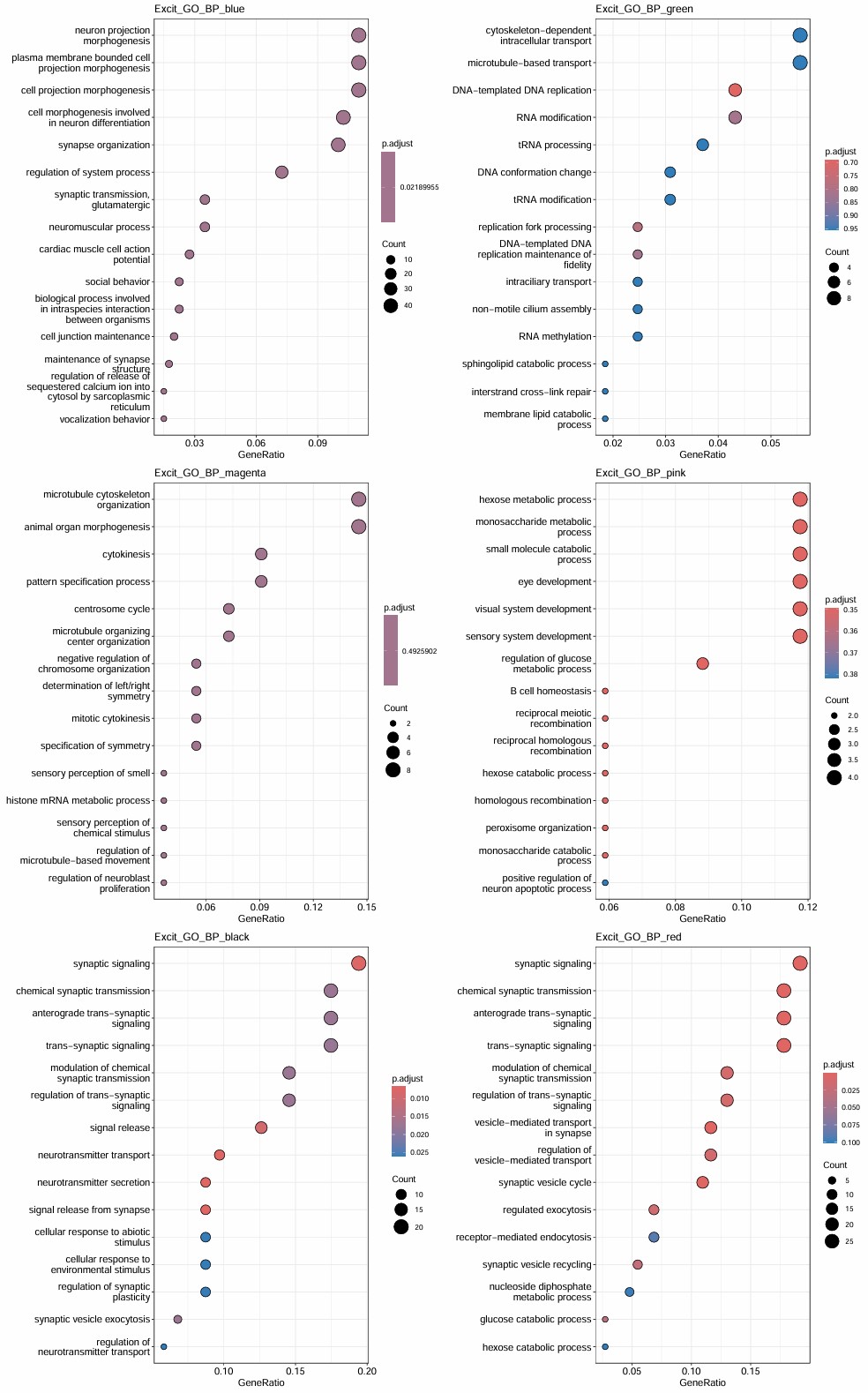
- 每个模块都有一些看起来和神经/免疫相关的,但都不太显著
- black和red和神经比较相关
- pink和免疫有关
- green和DNA/RNA有关
- 考虑到6在black和red下调,所以应该取6亚群下调基因与它们的交集
- 6有近3000个细胞,NC略多(1.4%),但其上调基因确实显著富集到神经相关条目上,black和pink模块与hERV负相关,6在这两个模块上调,且hERV表达量低
Inhib
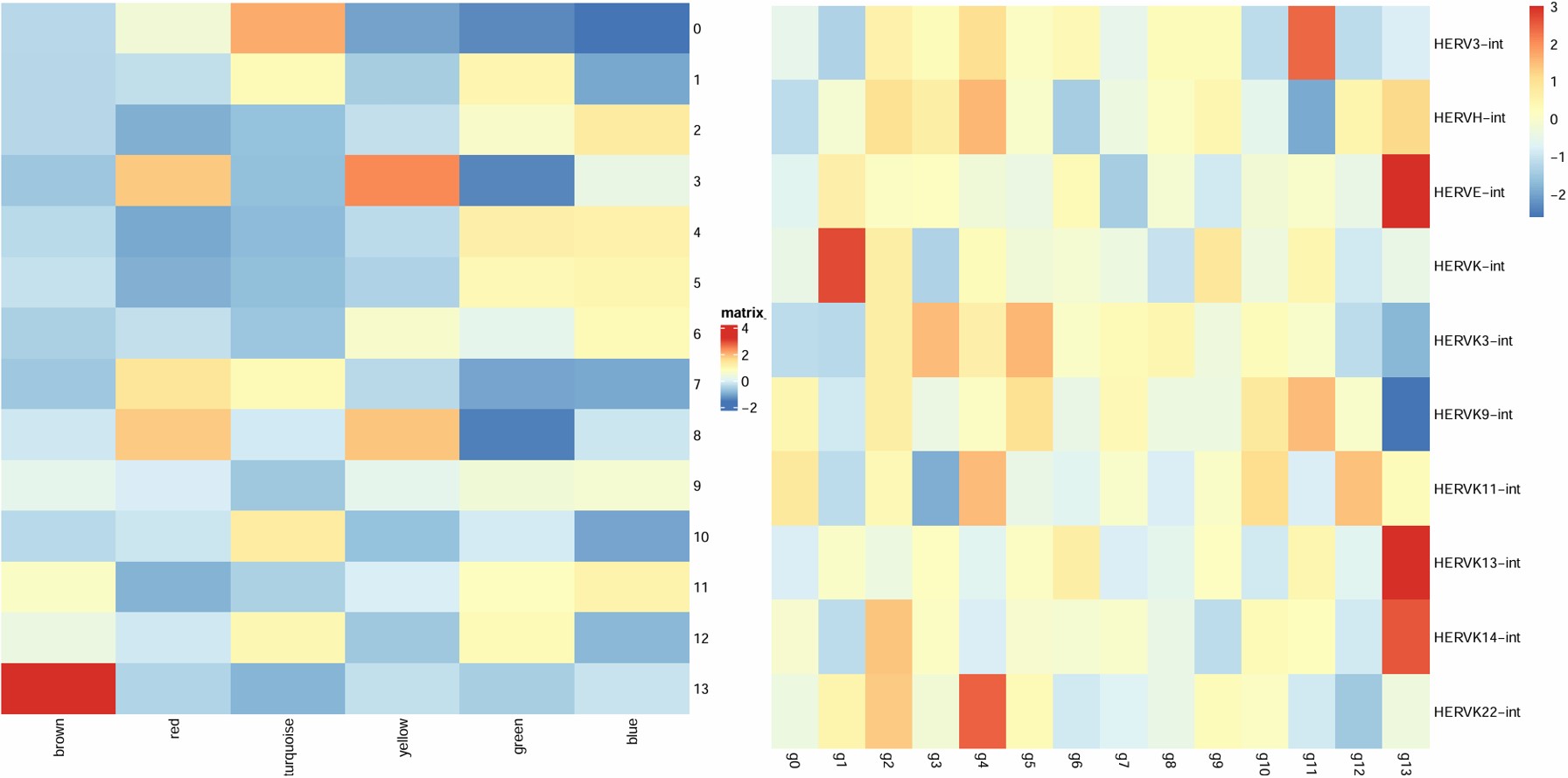
- 2没有特别负相关的模块,不过brown与hERV负相关,且2在其中表达很低,可以看看2和brown的关系
- 感觉没什么特别的
Endo
- 1在blue上调,red/turquoise下调
- 4在brown上调,yellow下调
- 主线应该就是1和blue,yellow相关性不太稳定
- blue基因数似乎太多了,感觉也没什么特别的
Mic
- blue和hERV显著负相关,3hERV表达高,但blue模块表达量也高,这对吗?
- 6在turquoise上调,brown/pink/black/red下调,然而turquoise/brown/pink/black/red都和hERV正相关,最主要6就100多个细胞,不太行
- 只有2有1000多个细胞,在blue微微下调,在turquoise/brown/pink微上调
- 如果mic很值得一看的话,大概就是blue/turquoise/brown/pink
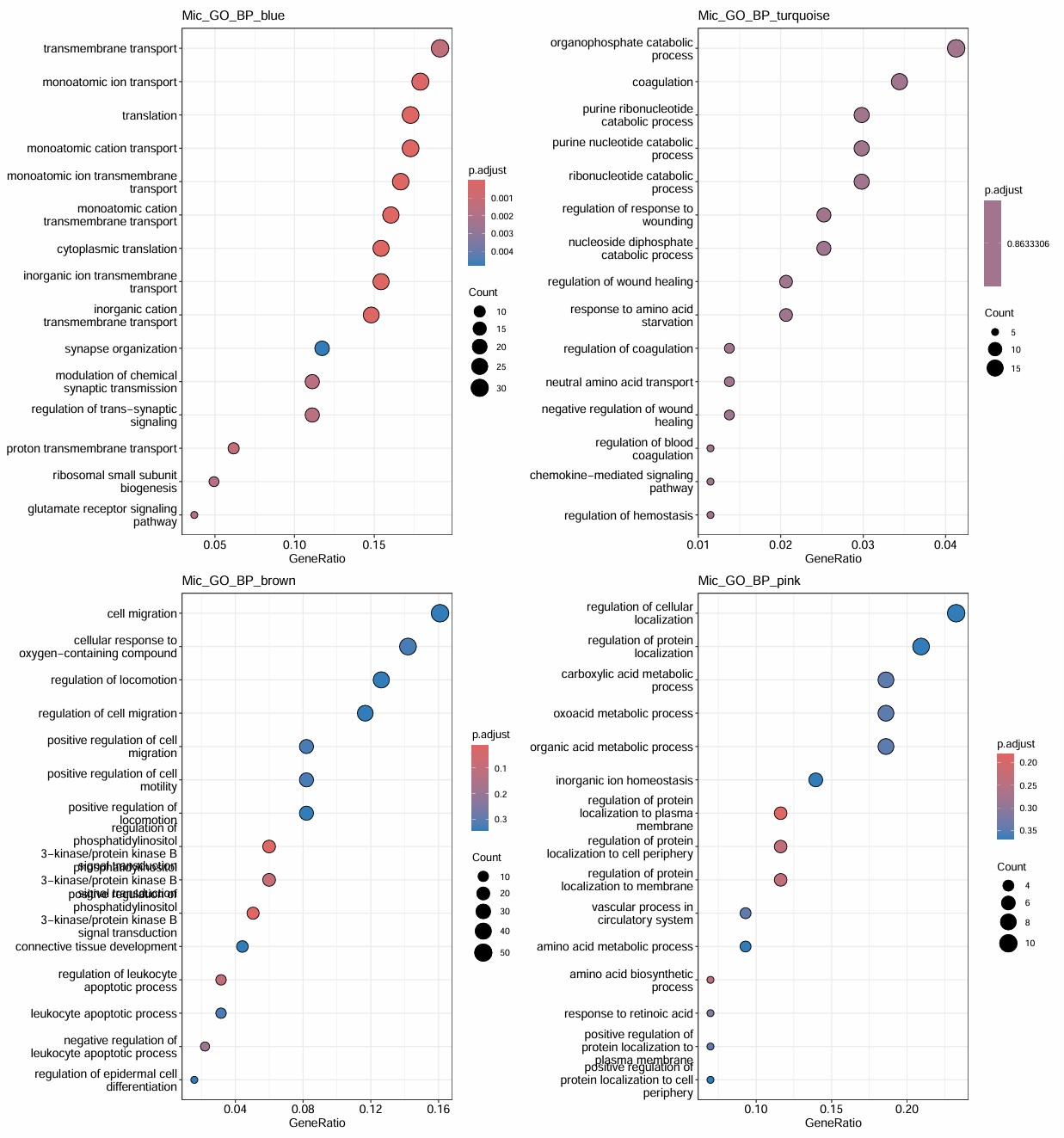
- blue的结构比较显著,与神经和翻译有相关,其它几个感觉有点普通了,可以先做个取交集看看
- 考虑到2在blue下调,所以应该取2亚群下调基因与它的交集
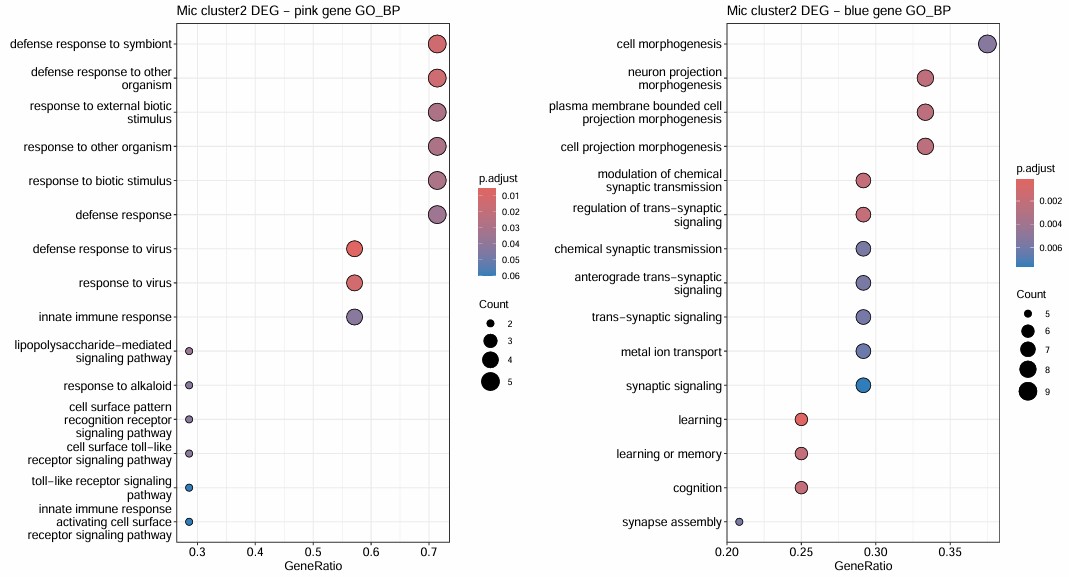
- 2AD多(13%),在blue这个神经相关模块下调,且hERVK等家族表达量高,hERVK与blue显著负相关;在pink这个信号响应?模块上调,pink与hERV正相关
-
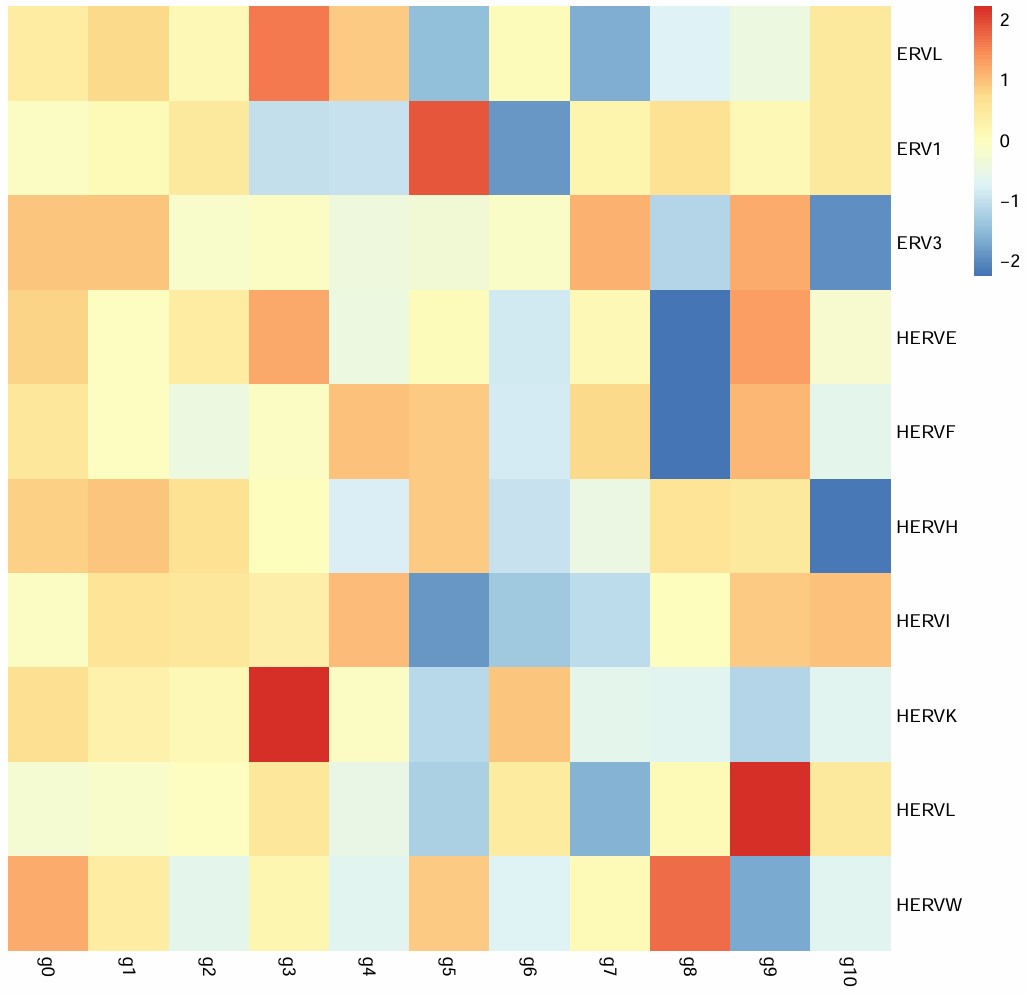
补一下mic的group热图,与hERVK的关系看得更明显
所有亚群的GO分析
对所有模块进行一遍GO分析,看有没有免疫相关模块、炎症相关模块(Mic),补充一下之前的结论
astro:也存在免疫/细胞因子响应方向的hERV关联,但强度和清晰度不如Mic,更像是“胶质支持/分化+轻度炎症响应”的背景状态
- blue:与hERV明显正相关,更偏向一个胶质分化/细胞黏附/神经支持相关状态
- green:与hERV也有正相关,TNF/cytokine response
- yellow:显著富集到蛋白质相关条目,与hERV有弱相关
- turquoise:与DNA修复有关,与hERV强正相关,但GO不显著
Oligo:hERV更稳定地关联到一个“广义信号调节/状态重塑”模块;但在具体亚群层面,某些hERV-high状态(比如cluster 9)又可以进一步落到“高翻译/高代谢/成熟髓鞘化”程序上
- turquoise和hERV相关性最强,GO条目分布比较广(该亚群基因数很多)
- green:成熟少突/髓鞘化模块,和hERV相关性一般
- brown:蛋白折叠/伴侣蛋白模块,和hERV相关性一般
Mic:hERV高表达更倾向于和先天免疫/炎症样程序增强相联系,同时与翻译/神经互作样程序下降相联系
- black:最像“先天免疫/抗病原体”模块,与hERV相关性较高
- pink:最像“炎症受体/信号响应”模块,虽然GO条目不是很集中,但hub gene中有TLR2/SP100/MAP3K8/EPSTI1,与炎症刺激/先天免疫信号响应有关
- yellow:蛋白折叠+免疫效应,可能说明Mic的hERV-high状态,可能不是单纯炎症,也可能伴随proteostasis/stress response
- blue:有cytoplasmic translation/synaptic signaling/glutamate receptor signaling/ribosome biogenesis条目,与hERV是负相关,可能说明Mic的hERV升高并不是简单伴随“全局活性增强”,而更像伴随某种homeostatic/neuron-interacting/translation-high状态的下降
Excit:hERV还可能与核酸代谢/复制样异常程序相关
- green模块的GO条目与DNA复制/核酸加工/应激样有关,且与hERV相关性较强,但GO不显著,也不是很标准的表观染色质调控模块
Inhib:与hERV相关性整体较弱,turquoise模块与hERV相关性较强,模块显著富集到突触相关条目,且与AD有弱负相关;brown与hERV有较强负相关,与蛋白折叠/应激有关,但GO不显著,可作为补充说明
Endo:与hERV相关性整体较弱,不过也有涉及到DNA合成/细胞分裂的yellow模块与hERV相关性较强,但GO不显著;brown显著富集到神经相关条目、green显著富集到蛋白质相关条目,但它们与hERV相关性较弱
有没有“STAT1等抗病毒通路相关的hERV”:
- STAT1指的是标准抗病毒程序:细胞感受到病毒核酸/dsRNA,从而激活免疫通路(type I / type II interferon)
- 上面提到的先天免疫/炎症样/抗原呈递样模块:细胞感受到损伤、病原相关分子、细胞碎片、炎症因子,通过TLR/NF-κB/MAPK/cytokine等通路进入激活状态,同时可能伴随吞噬、补体、抗原呈递等功能增强
- Mic中没有看到一个很典型的“STAT1-IFIT-OAS-MX1”式干扰素模块,有defense response/innate immune/TLR2/EPSTI1/MAP3K8(先天免疫/炎症样和抗原呈递样模块),不算特别像标准interferon/STAT1 signature
- OPC的green模块的hub gene里有STAT1,GO也有response to external biotic stimulus/response to other organism,与hERV也是正相关,但GO富集并不显著
总结byGPT
hERV关联不是统一的,而是明显细胞类型依赖:
- Astro、Oligo、OPC、Excit的模块-hERV相关性幅度更大
- Mic相关性中等,但模块的生物学解释很有意思
- Endo、Inhib整体较弱
AD和hERV不是简单同向:
- 很多模块里都能看到hERV和模块相关明显,但AD_binary相关很弱,甚至方向相反
- hERV更像是“细胞状态轴”的标志,而不是简单的AD/NC二分类标志
主线:
- Oligo:在整体层面,hERV更稳定地关联到“广义信号调节/状态重塑”模块。但在具体亚群层面,hERV高表达并不对应单一转录程序,而是分化为至少三类状态——一类偏膜脂与信号重塑,一类偏细胞骨架/黏附重塑,另一类则表现为成熟髓鞘化背景下的高翻译和高氧化磷酸化状态
- cluster 5:更像hERV激活型,偏脂质代谢、膜转运、信号/修饰重塑
- cluster 3:更像结构重塑/黏附变化/分化相关状态
- cluster 9:最清楚,GO直接落在cytoplasmic translation/oxidative phosphorylation/ATP metabolic process,这说明它是一个成熟髓鞘化、高代谢、高翻译活性的少突状态
- Mic:在整体层面,hERV高表达状态与抗病毒/先天免疫样反应增强相关,同时伴随翻译/神经互作相关程序的下降
- cluster 2:AD偏多(比全部Mic中的AD比例多13%),hERVK等家族偏高,与hERV正相关的pink模块上调,而pink模块富集到defense response to virus/response to virus/defense response to symbiont相关(DEG包括TLR2/MX2/MX1);与hERV负相关的blue模块下调,blue模块与learning/cognition/trans-synaptic signaling/neuron projection morphogenesis相关
- 除了pink和blue模块,在整体层面,Mic中的hERV与有关先天免疫/抗病原体的black模块、有关蛋白折叠和免疫效应的yellow模块有相关
支持性结果:
- Excit:提供了一个和神经胶质细胞不同的模式——是hERV较低的神经元状态,保留了更强的突触传递和囊泡循环程序;反过来说,hERV更多和激活/重塑/炎症类状态一起出现,hERV升高可能与神经元突触功能程序的减弱相关
- cluster 6的hERV表达量低,在red和black模块上调,而red和black的GO都是突触和信号传递相关的
- astro:hERV相关模块主要连接到gliogenesis、cell-cell signaling和synapse organization,提示其更可能反映胶质细胞参与神经支持和细胞间通讯的状态,也存在免疫/细胞因子响应方向的hERV关联,但强度和清晰度不如Mic,更像“胶质支持/分化+轻度炎症响应”的背景状态
总述:hERV重调在AD脑内具有明显的细胞类型和细胞状态依赖性。在不同细胞类型中,hERV并非统一对应同一种病理程序,而是分别耦联到不同的转录模块:在oligo中与膜脂重塑、细胞骨架重塑及成熟髓鞘化/高代谢状态相关;在mic中与广义先天免疫/炎症样激活、抗原呈递样程序增强相关,并与稳态和翻译程序下降相关;在astro中更多关联于胶质分化、神经支持与轻度细胞因子响应;而在excit中,hERV升高则更可能与突触传递程序减弱相联系。除此之外,在OPC中也存在一定STAT1/外源刺激响应线索,在inhib中与突触功能和蛋白折叠/应激有关,在endo中与DNA合成/细胞分裂、神经和蛋白质相关功能有关,但与hERV的相关性较弱或者是GO富集结果不显著,需要更多验证
Figure1&S1
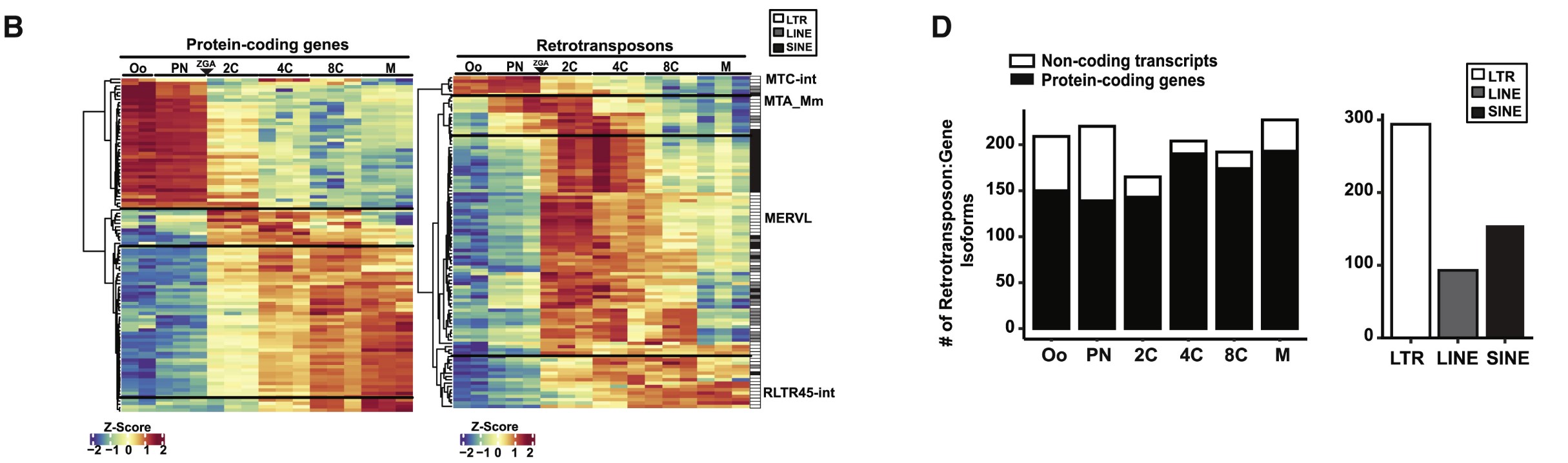
-
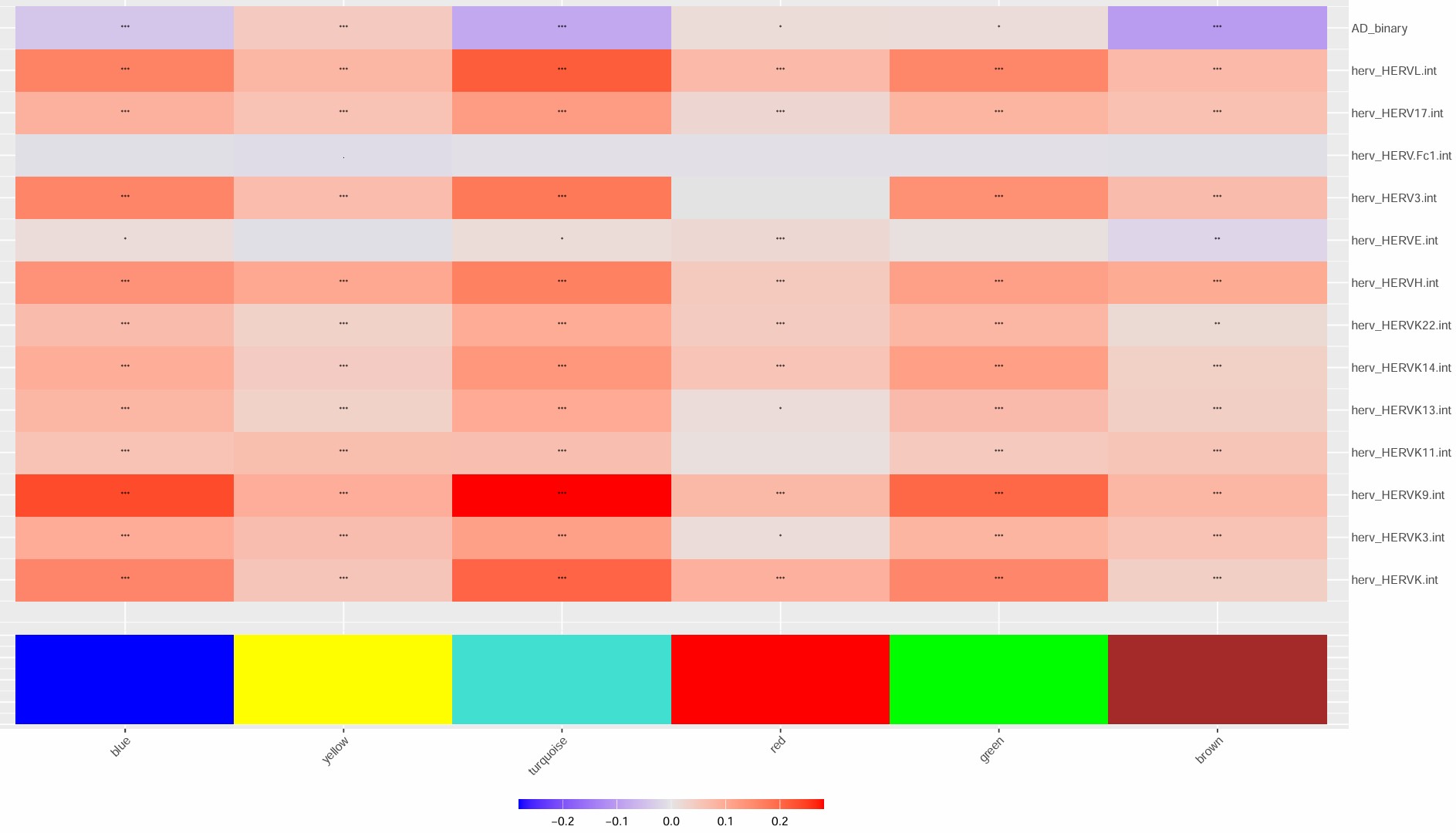
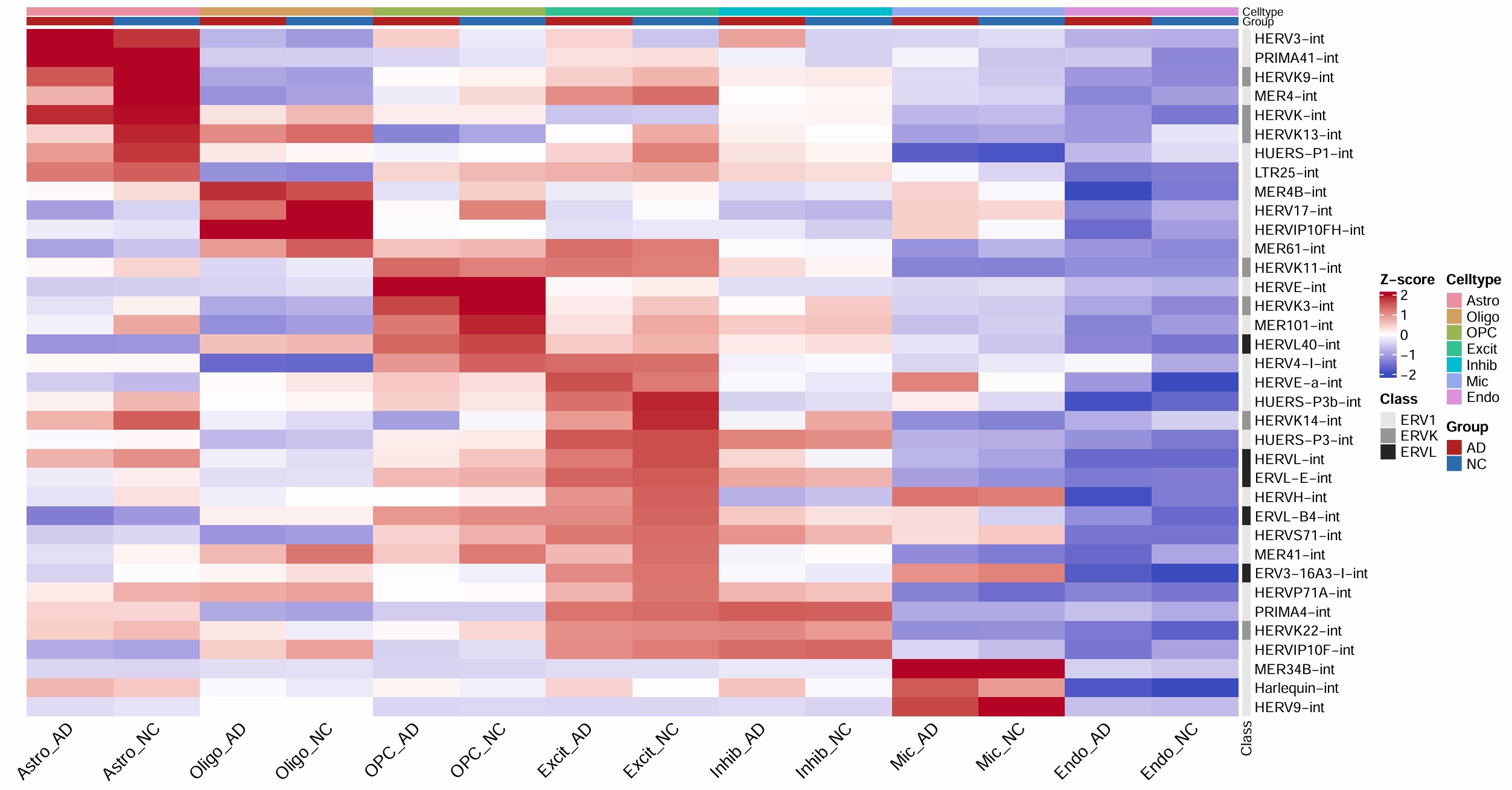
1B:在小鼠胚胎发育不同阶段中,前100个“高表达且动态变化明显”的蛋白编码基因/逆转座子家族,说明逆转座子的整体动态表达模式,与普通蛋白编码基因很相似,也随发育阶段发生有规律的切换
颜色不是绝对表达量,而是标准化后的相对高低(这个位点/家族在该列条件下相对它自己更高/低)
- 1D左:逆转座子-基因junction更常落在编码蛋白基因上,而不是非编码转录本上
- 1D右:这些逆转座子的类型
在我的研究中:只画热图,作为展示hERV整体分布的图。具体来说,把横轴的发育阶段换成Astro_NC/Astro_AD/Endo_NC/Endo_AD…,纵轴是是hERV家族(选一些高表达且在这些列之间变化较大的),右面加一个注释条标明超家族,先不做聚类,每个单元格的颜色表示该hERV家族在该细胞类型中的相对表达量,以此展示每个细胞类型中高表达的hERV有区别,经过测试,我选择了行标准化,即“同一个hERV家族在不同细胞类型中,哪里相对更高、哪里相对更低”(因为列标准化会出来很多hERV家族在所有细胞类型中表达都高,就没法体现出阶梯状的差异)
mat <- GetAssayData(seu, assay = "HERV_family", layer = "data")
celltype_order <- c("Astro", "Oligo", "OPC", "Excit", "Inhib", "Mic", "Endo")
group_order <- c("AD", "NC")
# 提取metadata,并构造列分组
meta <- seu@meta.data[, c("celltype", "group"), drop = FALSE]
mat <- mat[, rownames(meta), drop = FALSE]
meta$celltype <- factor(as.character(meta$celltype), levels = celltype_order)
meta$group <- factor(as.character(meta$group), levels = group_order)
mat <- mat[, rownames(meta), drop = FALSE]
meta$cellgroup <- paste(meta$celltype, meta$group, sep = "_")
col_order <- expand.grid(
celltype = celltype_order,
group = group_order,
stringsAsFactors = FALSE
)
col_order$cellgroup <- paste(col_order$celltype, col_order$group, sep = "_")
col_order <- col_order[order(
match(col_order$celltype, celltype_order),
match(col_order$group, group_order)
), , drop = FALSE]
col_order <- col_order[col_order$cellgroup %in% meta$cellgroup, , drop = FALSE]
# 计算每个celltype×group的平均表达
avg_list <- lapply(col_order$cellgroup, function(x) {
cells <- rownames(meta)[meta$cellgroup == x]
Matrix::rowMeans(mat[, cells, drop = FALSE])
})
avg_mat <- do.call(cbind, avg_list)
colnames(avg_mat) <- col_order$cellgroup
rownames(avg_mat) <- rownames(mat)
# 选择“高表达且变化较大”的 hERV family
row_mean <- rowMeans(avg_mat)
row_sd <- apply(avg_mat, 1, sd)
min_mean <- 0.05 # “高表达”的阈值
top_n <- 40 # 画多少个family
keep <- row_mean >= min_mean
stat_df <- data.frame(
family = rownames(avg_mat),
mean_expr = row_mean,
sd_expr = row_sd,
stringsAsFactors = FALSE
)
stat_df <- stat_df[keep, ]
stat_df <- stat_df[order(-stat_df$sd_expr, -stat_df$mean_expr), ] # 先按平均表达过滤,再按不同列之间的标准差排序
family_use <- head(stat_df$family, top_n)
plot_mat <- avg_mat[family_use, , drop = FALSE]
# 计算Z-score
plot_mat_z <- t(scale(t(as.matrix(plot_mat))))
plot_mat_z <- plot_mat_z[apply(plot_mat_z, 1, function(x) all(is.finite(x))), , drop = FALSE]
# 按“最高表达出现在哪一列”排序
peak_col <- max.col(plot_mat_z, ties.method = "first")
peak_val <- apply(plot_mat_z, 1, max)
ord <- order(peak_col, -peak_val)
plot_mat_z <- plot_mat_z[ord, , drop = FALSE]
# 加右侧超家族注释条
family_anno <- read.csv("/public/home/GENE_proc/wth/metadata/hERV/hERV_locus2family.csv")
family_anno$subfamily_raw <- gsub("_", "-", family_anno$subfamily_raw)
row_class <- family_anno$class[match(rownames(plot_mat_z), family_anno$subfamily_raw)]
row_class[is.na(row_class)] <- "Unknown"
class_cols <- c(
"ERV1" = "#E6E6E6",
"ERVK" = "#969696",
"ERVL" = "#252525",
"Unknown" = "#BDBDBD"
)
right_anno <- rowAnnotation(
Class = row_class,
col = list(Class = class_cols),
show_annotation_name = TRUE,
annotation_name_gp = gpar(fontsize = 10),
simple_anno_size = unit(2, "mm") # 右侧色条宽度
)
# 顶部注释
celltype_cols <- setNames(hcl.colors(length(celltype_order), "Set 2"), celltype_order)
group_cols <- c("NC" = "#2B6CB0", "AD" = "#B22222")
col_order$celltype <- factor(col_order$celltype, levels = celltype_order) # 固定图例标签顺序
col_order$group <- factor(col_order$group, levels = group_order)
top_anno <- HeatmapAnnotation(
Celltype = col_order$celltype,
Group = col_order$group,
col = list(
Celltype = celltype_cols,
Group = group_cols
),
annotation_legend_param = list(
Celltype = list(
at = celltype_order
),
Group = list(
at = group_order
)
),
annotation_name_gp = gpar(fontsize = 8),
simple_anno_size = unit(2, "mm") # 顶部色条高度
# , gap = unit(0.25, "mm") # 两条注释之间留一点小空隙
)
# 画图
ht <- Heatmap(
plot_mat_z,
name = "Z-score",
col = colorRamp2(c(-2, 0, 2), c("#3B4CC0", "white", "#B40426")),
top_annotation = top_anno,
right_annotation = right_anno,
cluster_rows = FALSE,
cluster_columns = FALSE,
show_row_names = TRUE,
show_column_names = TRUE,
row_names_gp = gpar(fontsize = 10),
column_names_gp = gpar(fontsize = 12),
column_names_rot = 45,
border = FALSE,
# column_title = "Average expression of hERV families",
# row_title = "hERV family",
rect_gp = gpar(col = NA) # 去掉单元格边框
)
pdf(file.path(res_dir, "hERV_family_heatmap.pdf"), width = 15, height = 8)
draw(ht, heatmap_legend_side = "right", annotation_legend_side = "right", padding = unit(c(5, 10, 5, 5), "mm"))
dev.off()
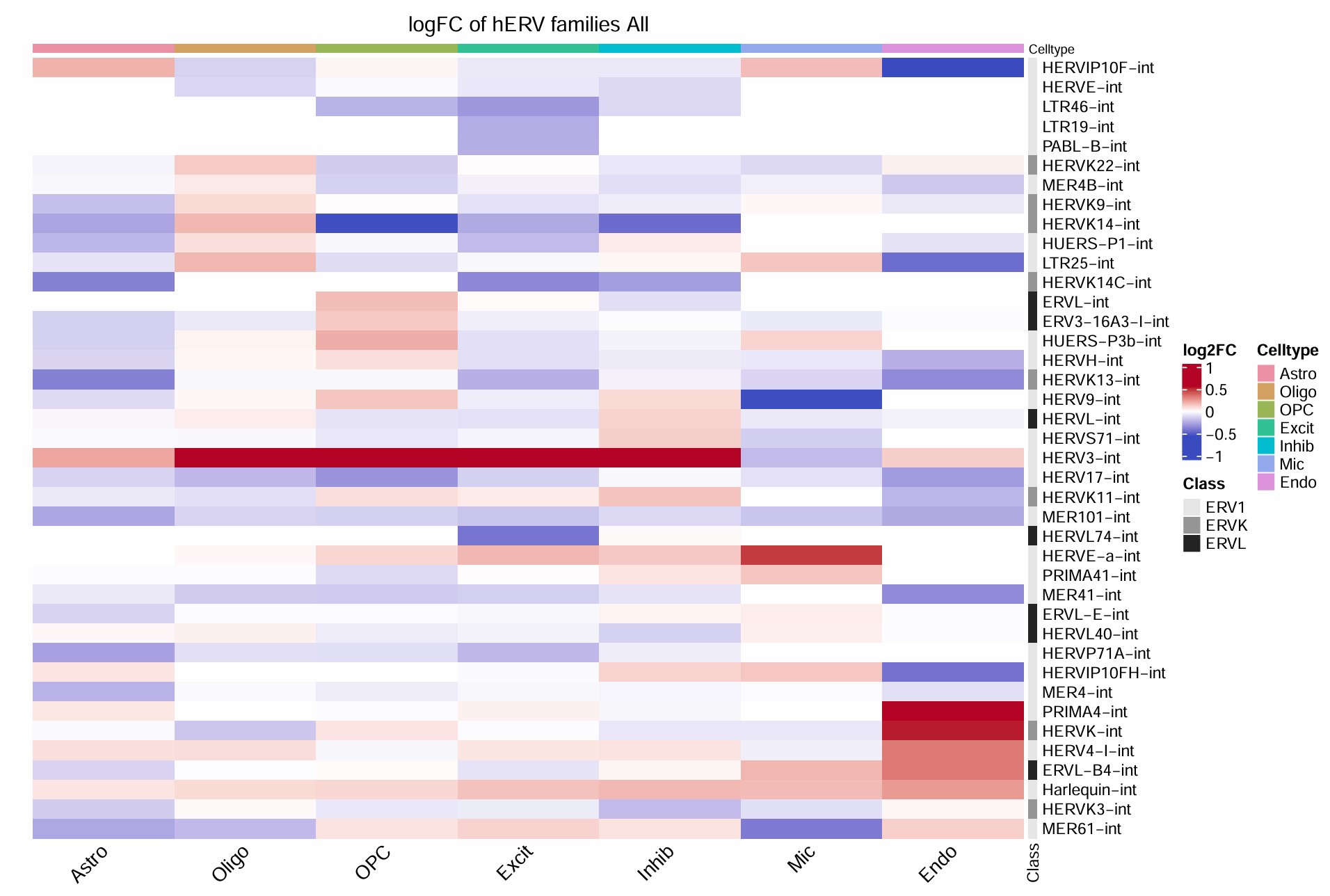
把z-score改成AD-NC的log2FC:即用hERV_family_celltype.csv画
de_df <- read_xlsx("C:\\Users\\17185\\Desktop\\hERV_AD_snRNA-seq_analyze\\res\\hERV_DE\\hERV_family_celltype.xlsx")
celltype_order <- c("Astro", "Oligo", "OPC", "Excit", "Inhib", "Mic", "Endo")
de_df <- de_df %>%
filter(celltype %in% celltype_order) %>%
select(celltype, subfamily, avg_log2FC)
logfc_df <- de_df %>%
pivot_wider(
names_from = celltype,
values_from = avg_log2FC
)
logfc_df <- logfc_df[, c("subfamily", celltype_order[celltype_order %in% colnames(logfc_df)])]
logfc_mat <- as.matrix(logfc_df[, -1])
rownames(logfc_mat) <- logfc_df$subfamily
logfc_mat[is.na(logfc_mat)] <- 0
row_sd <- apply(logfc_mat, 1, sd)
row_maxabs <- apply(abs(logfc_mat), 1, max)
stat_df <- data.frame(
family = rownames(logfc_mat),
sd_logFC = row_sd,
max_abs_logFC = row_maxabs,
stringsAsFactors = FALSE
)
stat_df <- stat_df[order(-stat_df$sd_logFC, -stat_df$max_abs_logFC), ]
top_n <- 40
family_use <- head(stat_df$family, top_n)
plot_mat <- logfc_mat[family_use, , drop = FALSE]
plot_mat_z <- t(scale(t(plot_mat)))
plot_mat_z[!is.finite(plot_mat_z)] <- 0
peak_col <- max.col(plot_mat_z, ties.method = "first")
peak_val <- apply(plot_mat_z, 1, max)
ord <- order(peak_col, -peak_val)
plot_mat <- plot_mat[ord, , drop = FALSE]
family_anno <- read.csv("C:\\Users\\17185\\Desktop\\hERV_AD_snRNA-seq_analyze\\metadata\\hERV\\hERV_locus2family.csv")
family_anno$subfamily_raw <- gsub("_", "-", family_anno$subfamily_raw)
row_class <- family_anno$class[match(rownames(plot_mat), family_anno$subfamily_raw)]
row_class[is.na(row_class)] <- "Unknown"
class_cols <- c(
"ERV1" = "#E6E6E6",
"ERVK" = "#969696",
"ERVL" = "#252525",
"Unknown" = "#BDBDBD"
)
right_anno <- rowAnnotation(
Class = row_class,
col = list(Class = class_cols),
show_annotation_name = TRUE,
annotation_name_gp = gpar(fontsize = 10),
simple_anno_size = unit(2, "mm")
)
celltype_cols <- setNames(hcl.colors(length(celltype_order), "Set 2"), celltype_order)
top_anno <- HeatmapAnnotation(
Celltype = factor(colnames(plot_mat), levels = celltype_order),
col = list(Celltype = celltype_cols),
annotation_legend_param = list(
Celltype = list(at = celltype_order)
),
annotation_name_gp = gpar(fontsize = 8),
simple_anno_size = unit(2, "mm")
)
lim <- quantile(abs(plot_mat), 0.98, na.rm = TRUE)
lim <- max(lim, 0.2)
ht <- Heatmap(
plot_mat,
name = "log2FC",
col = colorRamp2(c(-lim, 0, lim), c("#3B4CC0", "white", "#B40426")),
top_annotation = top_anno,
right_annotation = right_anno,
cluster_rows = FALSE,
cluster_columns = FALSE,
show_row_names = TRUE,
show_column_names = TRUE,
row_names_gp = gpar(fontsize = 10),
column_names_gp = gpar(fontsize = 12),
column_names_rot = 45,
border = FALSE,
column_title = "logFC of hERV families All",
# row_title = "hERV family",
rect_gp = gpar(col = NA),
na_col = "grey90",
)
组合图:
my_heatmap <- function(seu, de_res_path, herv_map_path = "/public/home/GENE_proc/wth/metadata/hERV/hERV_locus2family.csv"){
dataset <- identity_seu(seu, "dataset")
mat <- GetAssayData(seu, assay = "HERV_family", layer = "data")
celltype_order <- c("Astro", "Oligo", "OPC", "Excit", "Inhib", "Mic", "Endo")
group_order <- c("AD", "NC")
meta <- seu@meta.data[, c("celltype", "group"), drop = FALSE]
mat <- mat[, rownames(meta), drop = FALSE]
meta$celltype <- factor(as.character(meta$celltype), levels = celltype_order)
meta$group <- factor(as.character(meta$group), levels = group_order)
mat <- mat[, rownames(meta), drop = FALSE]
meta$cellgroup <- paste(meta$celltype, meta$group, sep = "_")
col_order <- expand.grid(
celltype = celltype_order,
group = group_order,
stringsAsFactors = FALSE
)
col_order$cellgroup <- paste(col_order$celltype, col_order$group, sep = "_")
col_order <- col_order[order(
match(col_order$celltype, celltype_order),
match(col_order$group, group_order)
), , drop = FALSE]
col_order <- col_order[col_order$cellgroup %in% meta$cellgroup, , drop = FALSE]
avg_list <- lapply(col_order$cellgroup, function(x) {
cells <- rownames(meta)[meta$cellgroup == x]
Matrix::rowMeans(mat[, cells, drop = FALSE])
})
avg_mat <- do.call(cbind, avg_list)
colnames(avg_mat) <- col_order$cellgroup
rownames(avg_mat) <- rownames(mat)
row_mean <- rowMeans(avg_mat)
row_sd <- apply(avg_mat, 1, sd)
min_mean <- 0.05
top_n <- 40
keep <- row_mean >= min_mean
stat_df <- data.frame(
family = rownames(avg_mat),
mean_expr = row_mean,
sd_expr = row_sd,
stringsAsFactors = FALSE
)
stat_df <- stat_df[keep, ]
stat_df <- stat_df[order(-stat_df$sd_expr, -stat_df$mean_expr), ]
family_use <- head(stat_df$family, top_n)
plot_mat <- avg_mat[family_use, , drop = FALSE]
plot_mat_z <- t(scale(t(as.matrix(plot_mat))))
plot_mat_z <- plot_mat_z[apply(plot_mat_z, 1, function(x) all(is.finite(x))), , drop = FALSE]
peak_col <- max.col(plot_mat_z, ties.method = "first")
peak_val <- apply(plot_mat_z, 1, max)
ord <- order(peak_col, -peak_val)
plot_mat_z <- plot_mat_z[ord, , drop = FALSE]
family_anno <- read.csv(herv_map_path)
family_anno$subfamily_raw <- gsub("_", "-", family_anno$subfamily_raw)
row_class <- family_anno$class[match(rownames(plot_mat_z), family_anno$subfamily_raw)]
row_class[is.na(row_class)] <- "Unknown"
class_cols <- c(
"ERV1" = "#E6E6E6",
"ERVK" = "#969696",
"ERVL" = "#252525",
"Unknown" = "#BDBDBD"
)
right_anno <- rowAnnotation(
Class = row_class,
col = list(Class = class_cols),
show_annotation_name = TRUE,
annotation_name_gp = gpar(fontsize = 0),
simple_anno_size = unit(2, "mm")
)
celltype_cols <- setNames(hcl.colors(length(celltype_order), "Set 2"), celltype_order)
group_cols <- c("NC" = "#2B6CB0", "AD" = "#B22222")
col_order$celltype <- factor(col_order$celltype, levels = celltype_order)
col_order$group <- factor(col_order$group, levels = group_order)
top_anno <- HeatmapAnnotation(
Celltype = col_order$celltype,
Group = col_order$group,
col = list(
Celltype = celltype_cols,
Group = group_cols
),
annotation_legend_param = list(
Celltype = list(
at = celltype_order
),
Group = list(
at = group_order
)
),
annotation_name_gp = gpar(fontsize = 0),
simple_anno_size = unit(2, "mm")
)
up <- Heatmap(
plot_mat_z,
name = "Z-score",
col = colorRamp2(c(-2, 0, 2), c("#3B4CC0", "white", "#B40426")),
top_annotation = top_anno,
right_annotation = right_anno,
cluster_rows = FALSE,
cluster_columns = FALSE,
show_row_names = TRUE,
show_column_names = FALSE,
row_names_gp = gpar(fontsize = 10),
column_names_rot = 45,
border = FALSE,
column_title = paste0("Average expression of hERV families - ", dataset),
column_title_gp = gpar(fontsize = 16),
rect_gp = gpar(col = NA)
)
de_df <- read.csv(de_res_path)
de_df <- de_df %>%
filter(celltype %in% celltype_order) %>%
select(celltype, subfamily, avg_log2FC)
logfc_df <- de_df %>%
pivot_wider(
names_from = celltype,
values_from = avg_log2FC
)
logfc_df <- logfc_df[, c("subfamily", celltype_order[celltype_order %in% colnames(logfc_df)])]
logfc_mat <- as.matrix(logfc_df[, -1])
rownames(logfc_mat) <- logfc_df$subfamily
row_sd <- apply(logfc_mat, 1, sd)
row_maxabs <- apply(abs(logfc_mat), 1, max)
stat_df <- data.frame(
family = rownames(logfc_mat),
sd_logFC = row_sd,
max_abs_logFC = row_maxabs,
stringsAsFactors = FALSE
)
stat_df <- stat_df[order(-stat_df$sd_logFC, -stat_df$max_abs_logFC), ]
top_n <- 40
family_use <- head(stat_df$family, top_n)
plot_mat <- logfc_mat[family_use, , drop = FALSE]
plot_mat_z <- t(scale(t(plot_mat)))
plot_mat_z[!is.finite(plot_mat_z)] <- 0
peak_col <- max.col(plot_mat_z, ties.method = "first")
peak_val <- apply(plot_mat_z, 1, max)
ord <- order(peak_col, -peak_val)
plot_mat <- plot_mat[ord, , drop = FALSE]
row_class <- family_anno$class[match(rownames(plot_mat), family_anno$subfamily_raw)]
row_class[is.na(row_class)] <- "Unknown"
right_anno <- rowAnnotation(
Class = row_class,
col = list(Class = class_cols),
show_annotation_name = TRUE,
annotation_name_gp = gpar(fontsize = 0),
simple_anno_size = unit(2, "mm")
)
celltype_cols <- setNames(hcl.colors(length(celltype_order), "Set 2"), celltype_order)
top_anno <- HeatmapAnnotation(
Celltype = factor(colnames(plot_mat), levels = celltype_order),
col = list(Celltype = celltype_cols),
annotation_legend_param = list(
Celltype = list(at = celltype_order)
),
annotation_name_gp = gpar(fontsize = 0),
simple_anno_size = unit(2, "mm")
)
lim <- quantile(abs(plot_mat), 0.98, na.rm = TRUE)
lim <- max(lim, 0.2)
down <- Heatmap(
plot_mat,
name = "log2FC",
col = colorRamp2(c(-lim, 0, lim), c("#3B4CC0", "white", "#B40426")),
top_annotation = top_anno,
right_annotation = right_anno,
cluster_rows = FALSE,
cluster_columns = FALSE,
show_column_names = FALSE,
show_row_names = TRUE,
row_names_gp = gpar(fontsize = 10),
border = FALSE,
column_title = paste0("logFC of hERV families - ", dataset),
column_title_gp = gpar(fontsize = 16),
rect_gp = gpar(col = NA),
na_col = "grey90",
)
up <- as.ggplot(up)
down <- as.ggplot(down)
pdf(file.path(res_dir, paste0("hERV_family_heatmap_", dataset, ".pdf")), width = 10, height = 12)
print(up / down)
dev.off()
}
其它问题
dotplot表达量负数
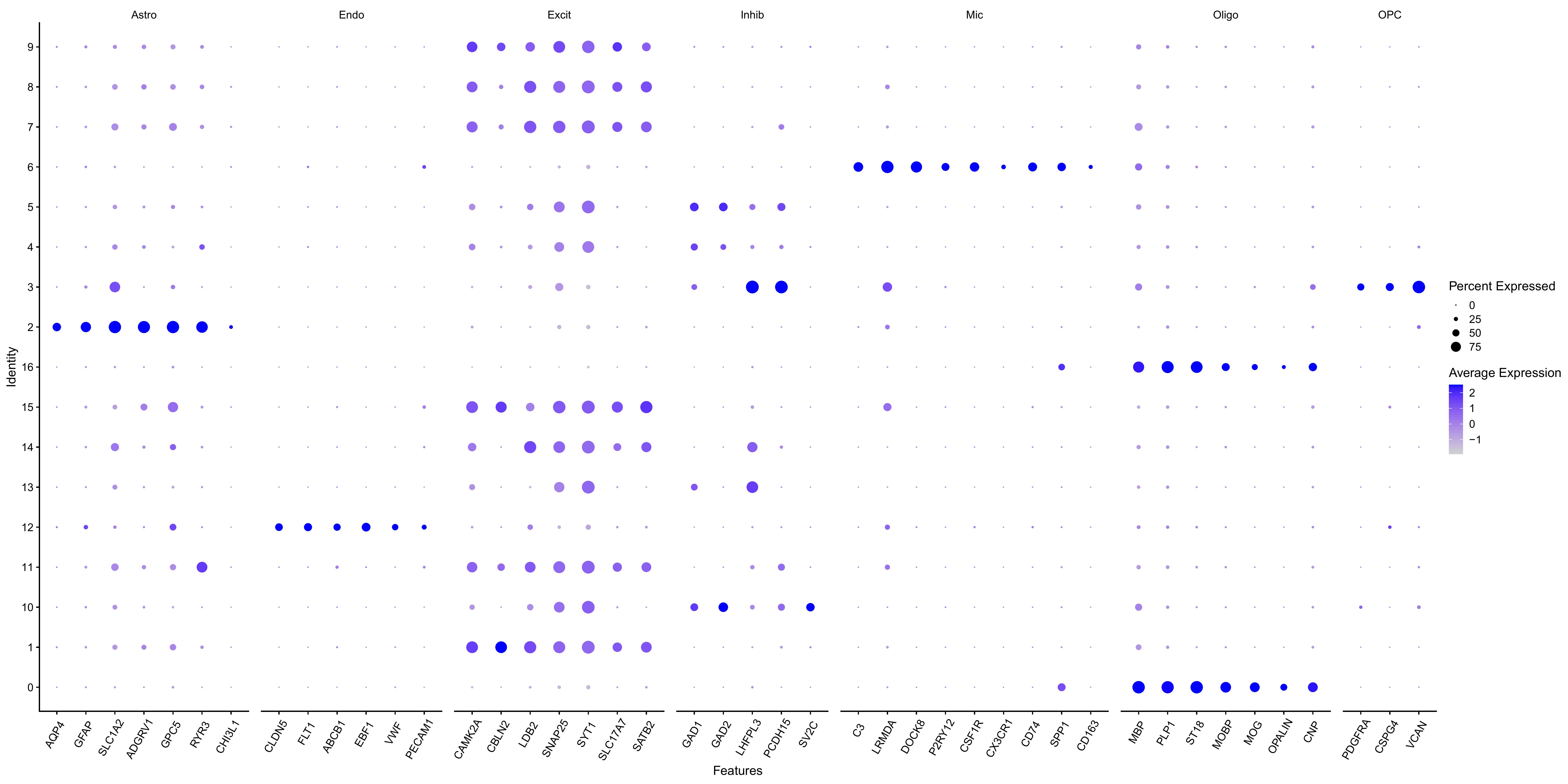
Average Expression:经过标准化后的平均表达量。DotPlot默认会对每个基因在不同细胞群之间的平均表达做缩放,缩放之后,数值表示的是这个基因在某个群里的表达,相对于该基因在所有群中的整体水平是高还是低:
- 大于0:这个群里该基因表达高于该基因的整体平均水平
- 小于0:这个群里该基因表达低于该基因的整体平均水平
- 0附近:接近平均水平
如果在DotPlot设置参数scale = FALSE,就是原始平均表达量
测序方法对hERV计数的影响
使用的数据的建库方法是10x Chromium Single Cell 3′ v3——“用poly(dT)引物捕获带poly(A)尾的转录本”,可能会低估或者漏掉不带polyA的ncRNA(非编码RNA),同时TE里面有相当一部分是non-polyA的,所以在hERV计数上大概率会偏低。不过我这个分析主要是AD-NC/不同细胞类型之间的组间比较,或者是共表达这种定性的找趋势,所以在这类不需要精确计数的分析上应该影响不大。当然也不可能说完全没有影响:
- 之前别人的bulk RNA-seq分析用的是total RNA的测序方法(而不是我这种polyA+的测序方法),这可能是结论有差异的原因之一
- 我这里能被polyA+的测序方法测出来的hERV读段,可能是hERV中有一定特殊性的片段,比如是剪接到了正常gene中的、或是借用了周围的polyA
hERV被基因内化为外显子或是启动子/增强子对测序的影响:
- 内化为外显子:更偏向polyA+,因为这时它本质上已经成了宿主基因转录本的一部分,后续剪接、加polyA、输出等过程大多按宿主mRNA或某些lncRNA的规则来走
- 内化为替代启动子:也常常更偏向polyA+,因为很多情况下是 LTR 驱动了一个宿主基因的替代转录本,这个转录本后面接宿主外显子,最后通常会形成比较稳定的加polyA尾的RNA
- 内化为增强子:更偏向non-polyA,因为增强子大多数不产生稳定RNA
古老型hERV更容易被基因内化为外显子或是启动子/增强子?
- 总体上,较老的TE/ERV更容易在进化中被宿主共选为调控元件。比如hERVK(HML-2)属于hERV中较年轻、最接近仍保留病毒样转录潜力的家族之一,而hERVL/ERVL就属于更古老的谱系,但在同一家族内部,也会有一些特殊的位点(比如hERVH就有一些年轻的高转录位点)
- hERV的年龄更可能改变“它以什么形式被转录出来”,而不是直接规定它一定是polyA还是non-polyA
- 古老型如果更多作为外显子/启动子(被内化进宿主基因转录本),polyA+比例会上升
- 古老型如果更多作为增强子产生调控RNA,non-polyA比例会上升
- 年轻型如果更多保持自主病毒样转录,polyA+也可能很高
- polyA测序更容易检测到
- 嵌在宿主polyA转录本里的hERV片段
- 本身能形成polyA尾的hERV转录本
- 从热图上看,也是既有ERVL/HERVL,也有hERVK这类,似乎并没有说高表达的更偏向年轻/古老
Human hippocampal neurogenesis
Human hippocampal neurogenesis in adulthood, ageing and Alzheimer’s disease
主要结论:成人海马神经发生是存在的;AD不只是“新生神经元少了”,更像是从前体细胞开始,整条神经发生轴线的表观调控网络逐步失稳;而认知保持得特别好的人,保留了一套与这种失稳相反的“韧性网络”
- 人类成年海马确实存在一条可识别的神经发生轨迹:用snRNA-seq和snATAC-seq联合分析,识别出了NSCs(神经干细胞)、neuroblasts(神经母细胞)、immature neurons(未成熟神经元),并且通过RNA velocity和调控网络分析支持它们构成一条从干/祖细胞到未成熟神经元的连续轨迹
- 随着衰老和AD进展,最稳定、最明显的变化不是RNA,而是染色质开放性:与DEGs相比,DARs(差异染色质开放区域)的变化更多、更强,也更能区分正常衰老、临床前阶段和AD。尤其在PCI(可理解为向AD过渡的临床前病理状态)里,一些和突触可塑性、神经传递、神经元结构维护有关的开放染色质区域已经开始下调,而这些变化在AD中更严重
- 最早的异常更可能起始于NSC,后面在neuroblast和immature neuron里放大
- SuperAgers(在情景记忆测试中表现出色的老年人)有更独特的neurogenesis profile,尤其是在immature neurons/neuroblasts上更明显,而且这种“resilience signature”主要也是体现在开放染色质和调控网络上,这种韧性与海马内astrocytes、CA1 neurons、谷氨酸能突触通路之间的相互作用有联系,“认知功能未明显衰退”和“认知衰退”分岔点可能就在这些细胞间调控网络上
TE和免疫系统
Transposable elements as instructors of the immune system
主要结论:TE在多个层面作为免疫系统行为的调控元件
- TE被转录后产生RNA、cDNA、蛋白或肽段,这些产物能被先天免疫识别,或者被MHC呈递给T细胞
- TE还能作为顺式调控元件,重塑某些免疫细胞的基因表达程序,或充当某些免疫细胞状态转换和组织适应程序中的增强子:免疫系统可能反复利用TE作为调控方式
- TE转录出来的RNA或RNA-DNA产物可以激活RIG-I、MDA5、cGAS–STING等先天免疫通路,从而诱导I型干扰素反应
- 在T细胞里,某些含LINE-1的转录本和T细胞的活化、分化、耗竭状态有关
- 在胸腺里,TE来源的转录本和肽段甚至可能参与中枢耐受,防止免疫系统攻击自身正常细胞
- 某些ERV类TE影响辅助性T细胞分化
- 在不同组织免疫细胞的scATAC-seq里找到了一批与组织适应有关的TE家族/亚家族,发现这些TE所在开放染色质里富集了BATF、JUNB等关键转录因子结合位点
- 未来可能用于癌症免疫治疗,也可能用于炎症和自身免疫病的干预
TE增强子调控网络如何计算:
- 先找候选增强子:用ATAC-seq或scATAC-seq找开放染色质区域
- 把开放峰和TE注释重叠:把ATAC峰和RepeatMasker之类的TE注释表相交,筛出“落在TE上的开放峰”,这些就是候选的TE-derived enhancers。然后再统计哪些TE家族/亚家族在某个细胞状态中显著富集
- motif enrichment推断谁在用这些TE:如果某些开放的TE峰里富集BATF、JUNB、TH1相关TF(转录因子)的motif(序列中的特定模式),就提示这些TF可能通过这些TE位点来驱动该细胞程序
- 把峰连到基因:做peak-gene correlation、co-accessibility,或者用同一细胞的RNA+ATAC多组学数据去关联“这个开放峰变化”和“哪个基因表达变化同步”
- 把这些关系拼成网络,并做实验验证
JETs(外显子和TE序列拼接形成的非经典转录本)在肿瘤里已经显示出很有意思的抗原潜力:一些肿瘤特异的JETs可以被翻译成蛋白,并作为肽段经MHC-I呈递,还能诱导T细胞反应。虽然他还没有被证实为神经系统疾病标志物,不过潜力很大