使用我的GSE157827数据进行初步分析-5
other-other2026.3.18-2026.3.25研究进展
GSE174367
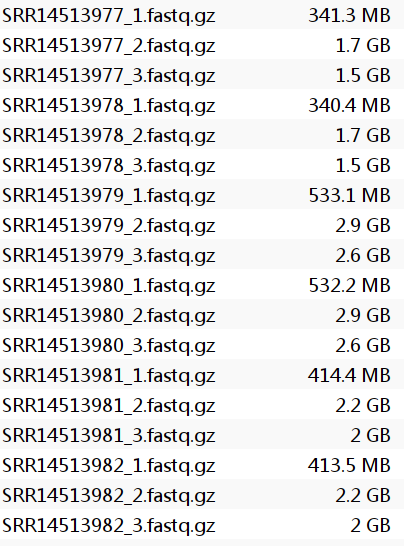
先看这几个1/2/3都是什么部分:
cd /public/home/GENE_proc/wth/GSE174367/fastq/
for fq in SRR14513977_{1,2,3}.fastq.gz; do
echo "===== $fq ====="
zcat "$fq" | awk '
NR%4==2 && ++n<=200000 {len[length($0)]++}
END{
for (l in len) print l, len[l]
}' | sort -n
echo
done
SRR14513977_1.fastq.gz:都是8bp,是sample index read- 正常来讲另外两个应该一个是28bp,另一个是90-100bp,但很神奇的是另外两个都是100bp
- GPT说是没切成28bp的形式,而是原始格式,需要自己看一下哪个是barcode+UMI,哪个是cDNA
import gzip
import sys
import math
from collections import Counter
N = 200000
MAX_POS = 40
def rc(seq):
comp = str.maketrans("ACGTN", "TGCAN")
return seq.translate(comp)[::-1]
def iter_seqs(fq, n=N):
count = 0
with gzip.open(fq, "rt") as f:
for i, line in enumerate(f):
if i % 4 == 1:
yield line.strip()
count += 1
if count >= n:
break
def entropy(counter):
total = sum(counter.values())
if total == 0:
return 0.0
h = 0.0
for v in counter.values():
p = v / total
h -= p * math.log2(p)
return h
def load_whitelist(path):
wl = set()
opener = gzip.open if path.endswith(".gz") else open
with opener(path, "rt") as f:
for line in f:
wl.add(line.strip())
return wl
def analyze_fastq(fq, whitelist=None):
seqs = list(iter_seqs(fq, N))
if not seqs:
print(f"\n===== {fq} =====")
print("No reads found")
return
lens = Counter(map(len, seqs))
dom_len, dom_n = lens.most_common(1)[0]
print(f"\n===== {fq} =====")
print(f"sampled reads: {len(seqs)}")
print("length distribution:")
for l, c in sorted(lens.items()):
print(f" {l} bp\t{c}")
print(f"dominant length: {dom_len} bp ({dom_n/len(seqs):.2%})")
p16 = [s[:16] for s in seqs if len(s) >= 16]
p28 = [s[:28] for s in seqs if len(s) >= 28]
c16 = Counter(p16)
c28 = Counter(p28)
print(f"unique first16: {len(c16)} / {len(p16)} ({len(c16)/len(p16):.2%})")
print(f"unique first28: {len(c28)} / {len(p28)} ({len(c28)/len(p28):.2%})")
print("top 10 first16:")
for s, c in c16.most_common(10):
print(f" {c}\t{s}")
print("top 10 first28:")
for s, c in c28.most_common(10):
print(f" {c}\t{s}")
if whitelist is not None:
hit = sum((x in whitelist) for x in p16)
hit_rc = sum((rc(x) in whitelist) for x in p16)
print(f"whitelist hit rate (first16): {hit/len(p16):.2%}")
print(f"whitelist hit rate (reverse-complement first16): {hit_rc/len(p16):.2%}")
tails = [s[28:60] for s in seqs if len(s) >= 60]
polyA = sum(("AAAAAAAAAAAA" in x) for x in tails)
polyT = sum(("TTTTTTTTTTTT" in x) for x in tails)
print(f"reads with >=12A in pos29-60: {polyA/len(tails):.2%}")
print(f"reads with >=12T in pos29-60: {polyT/len(tails):.2%}")
pos_counts = [Counter() for _ in range(MAX_POS)]
for s in seqs:
for i, b in enumerate(s[:MAX_POS]):
pos_counts[i][b] += 1
print("\nPer-cycle summary (first 40 cycles):")
print("pos\tmajor_base\tmajor_frac\tentropy\tA\tC\tG\tT\tN")
for i, cnt in enumerate(pos_counts, start=1):
total = sum(cnt.values())
if total == 0:
continue
major_base, major_n = cnt.most_common(1)[0]
A = cnt.get("A", 0) / total
C = cnt.get("C", 0) / total
G = cnt.get("G", 0) / total
T = cnt.get("T", 0) / total
Nn = cnt.get("N", 0) / total
H = entropy(cnt)
print(f"{i}\t{major_base}\t{major_n/total:.3f}\t{H:.3f}\t{A:.3f}\t{C:.3f}\t{G:.3f}\t{T:.3f}\t{Nn:.3f}")
if __name__ == "__main__":
if len(sys.argv) < 3:
print("Usage: python inspect_10x_cb_umi.py whitelist.txt.gz file1.fastq.gz file2.fastq.gz ...")
sys.exit(1)
wl = load_whitelist(sys.argv[1])
for fq in sys.argv[2:]:
analyze_fastq(fq, wl)
cd /public/home/GENE_proc/wth/GSE174367/fastq/
python ../inspect_10x_cb_umi.py \
/public/home/wangtianhao/Desktop/STAR_ref/whitelist/3M-february-2018.txt \
SRR14513977_2.fastq.gz \
SRR14513977_3.fastq.gz
最后算出来_2的前16bp对whitelist的命中率高达96%,并且它的unique first16只有14.52%,但加上12bp以后,unique first28达到91.62%,说明前16位是barcode,后12bp是UMI
先跑一个测试:
module load miniconda3/base
conda activate STAR
cd /public/home/GENE_proc/wth/GSE174367/
STAR \
--runMode alignReads \
--runThreadN 8 \
--genomeDir /public/home/wangtianhao/Desktop/STAR_ref/hg38/ \
--readFilesIn fastq/SRR14513977_3.fastq.gz fastq/SRR14513977_2.fastq.gz \
--readFilesCommand zcat \
--outFileNamePrefix star_test/SRR14513977 \
--soloType CB_UMI_Simple \
--soloCBstart 1 \
--soloCBlen 16 \
--soloUMIstart 17 \
--soloUMIlen 12 \
--soloBarcodeReadLength 0 \
--soloCBwhitelist /public/home/wangtianhao/Desktop/STAR_ref/whitelist/3M-february-2018.txt \
--soloFeatures GeneFull \
--clipAdapterType CellRanger4 \
--soloCellFilter EmptyDrops_CR \
--soloCBmatchWLtype 1MM_multi_Nbase_pseudocounts \
--soloUMIfiltering MultiGeneUMI_CR \
--soloUMIdedup 1MM_CR
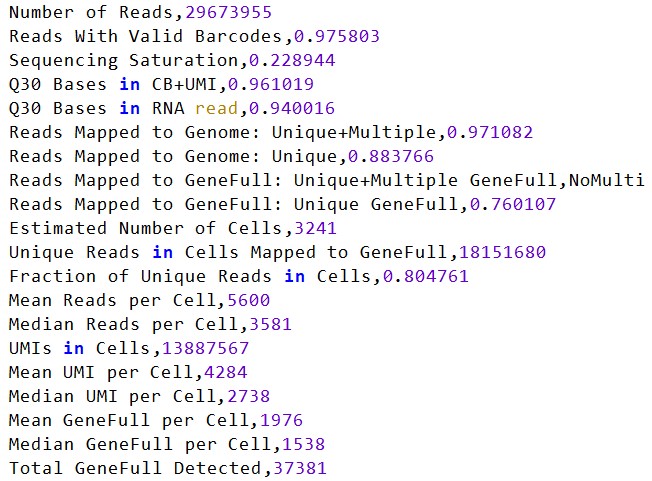
再跑一个完整的样本(包含多个SRR)
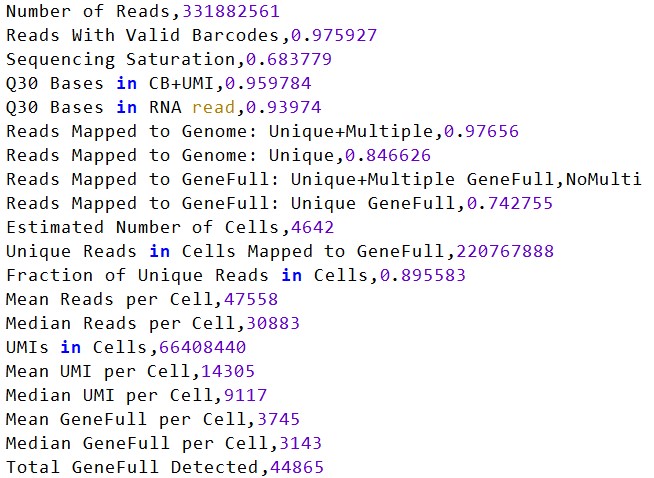
这个GSM5292838在作者的统计表里有4605个细胞(我的有4642个细胞),可以说是完全吻合,由此这个计数参数应该是没有什么问题的