暑假前文献阅读和计划安排
other-other暑假前文献阅读和计划安排
文献阅读
Integrated multimodal cell atlas of Alzheimer’s disease
- 全球首个阿尔茨海默病多模态细胞图谱
- 如果HERV在一个基因内部(同义链),且基因本身也表达,则有可能是构成了这个基因转录本的一个Isoform; 如果该基因表达低、HERV在反义链、或HERV周围没有基因,则更可能是它自己的转录
- 这些microglia-like cells(小胶质样脑巨噬细胞)跟你在mic里看到的亚组有关嘛
stellarscope原文:A single-cell transposable element atlas of human cell identity
- 用hERV定义细胞类型
- 分析非常全面 可以效仿 就是生物学上差点意思
2025 NG–A retrotransposon caught red-handed in a curious case of missing digits
- 这篇论文提到了一个比较有趣的方法,用现有的单细胞大模型Geneformer打底,用特定的case/control数据做微调,然后做in silico perturbation找关键基因。可学习下这个思路
Classification and characterization of human endogenous retroviruses; mosaic forms are common
- 近期重磅论文比如cell等里引用参考的HERV的分类
包含脑在内的单细胞atac:https://health.tsinghua.edu.cn/human-scatac-corpus/download.php
- 单细胞的eqtl,也许用得上
Spatiotemporal transcriptome atlas of human embryos after gastrulation
- 可以看看有没有fastq或bam可以用来找HERV 脑的数据能不能分得出
Activation of endogenous retroviruses during brain development causes an inflammatory response
- Cre诱导的Trim28在脑发育过程中神经细胞中丧失,导致成人大脑中ERV表达
2024 NC–hERV + TWAS
样本情况:CommonMind Consortium(CMC)数据集,人类死后脑的背外侧前额叶皮层(DLPFC)
- 初始样本:910个,表达数据、基因型数据、临床信息,包含无精神疾病诊断442人、精神分裂症350人、双相情感障碍110人
- 基因型数据:SNP基因型矩阵,比如每个人在某个位点是AA、AG还是GG
- 选择了两个较大的、相对同质的祖源群体:欧洲血统共563人+非洲血统共229人,都是包含患病和健康
- “把死亡时有精神疾病诊断的个体也纳入SNP权重构建,是因为增加样本量能提高检测cis调控效应的能力”
- “结果显示,使用完整欧洲样本比只用242个未受影响个体更有统计功效,且两者rTWAS结果高度一致”
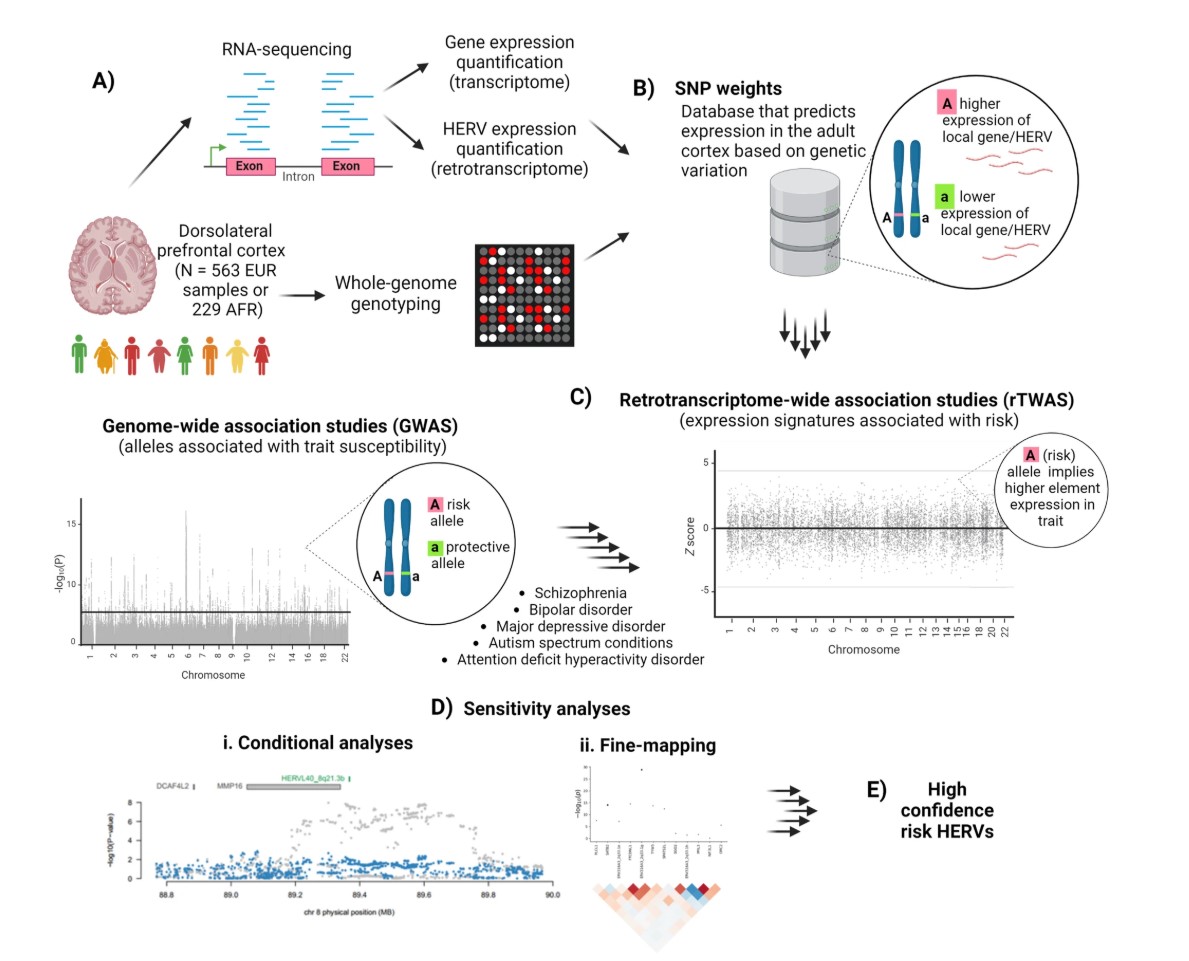
数据预处理:
-
基因型QC:去除了杂合率异常的样本、亲缘关系较高的样本、基因型缺失率较高的样本,以及遗传性别和记录性别不一致的样本
- 杂合率异常:可能提示样本污染、测序/芯片质量问题或群体结构异常
- pihat>0.2:PLINK中估计的样本亲缘关系指标,>0.2说明两个人可能有较近亲缘关系。TWAS建模一般希望样本近似独立
- fastq处理:Trimmomatic去除低质量碱基和过短reads
第1步:在DLPFC脑组织中定量gene和HERV表达
- 位点级HERV表达定量:Bowtie2 + Telescope
-
常规基因表达定量:
- kallisto不做传统逐碱基全比对,而是pseudoalignment,速度快,适合转录本/基因表达定量
- lengthScaledTPM是tximport中常用设置,用TPM回推count,同时考虑长度校正,方便后续统计模型使用
- biomaRt用于把Ensembl等数据库注释转换成标准gene信息
- 作者把有HUGO Gene Nomenclature Committee gene symbol的基因定义为canonical genes(常规基因/标准基因),主要是蛋白编码基因
-
表达过滤:把HERV和gene表达矩阵合并,然后只保留在至少20%样本中满足read counts≥6且TPM≥0.1的feature
- 这个阈值来自GTEx eQTL分析习惯,目的是去掉极低表达、难以可靠建模的feature。对于TWAS来说,表达太稀疏的feature无法稳定估计SNP weights
-
校正协变量:用limma调整表达数据,协变量包括样本来源机构、病例/对照状态、RIN、性别、死后间隔、年龄分组、前10个祖源PC,以及sva计算的surrogate variables
- limma:常用于表达矩阵线性模型校正
- RNA完整性指标(RIN):死后脑RNA质量差异很大,必须校正
- 死亡到取材间隔(post-mortem interval, PMI):会影响RNA降解
- 祖源主成分(population PCs):用于控制群体结构
- 未知批次效应或隐含混杂因素(surrogate variables, sva):例如实验批次、RNA质量残留差异、未记录的技术因素等
- 年龄分箱:作者把年龄分为17–29、30–49、50–69、70–89、90+,而不是直接使用连续年龄,可能是为了处理非线性年龄效应和>90岁这种顶码记录
第2步:构建SNP weights(遗传变异预测表达)模型
- Fig.1B:在某个局部基因组区域中,如果某个SNP的A等位基因对应某个gene/HERV更高表达,那么就可以学习出这个SNP对表达的权重。“用附近SNP预测某个gene/HERV表达量”
-
FUSION TWAS权重计算:在欧洲/非洲CMC子集中分别估计每个gene/HERV的cis遗传调控模型,Expressiongene/HERV ≈ β1SNP1 + β2SNP2 + … + βkSNPk,其中这些SNP都在该feature附近1Mb内
- 其中使用了BLUP/BSLMM/LASSO/Elastic net/Top SNP模型,这些模型对应不同遗传架构假设。某个HERV表达可能由一个强SNP控制,也可能由多个弱SNP共同控制。FUSION会尝试多种模型,从而提高不同类型feature的预测能力
- eQTL分析通常逐个SNP-feature配对检验,而TWAS权重构建更像是构建一个由多个cis SNP共同预测表达的模型
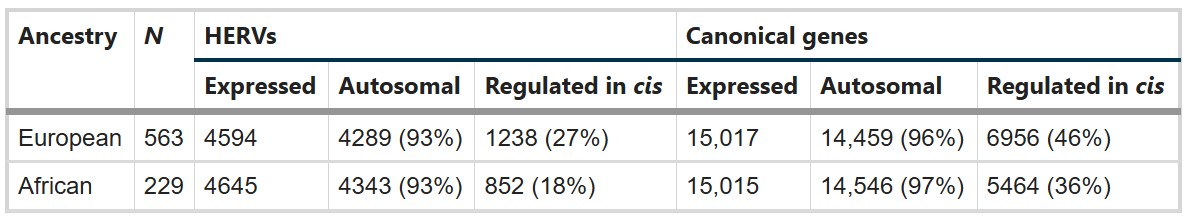
- 在DLPFC中能检测到多少HERV?其中多少有cis遗传调控?
- 欧洲血统563个样本中,检测到4594个表达HERV,其中4289个位于常染色体,1238个有显著cis遗传调控;有15017个表达基因,6956个有cis调控
- 非洲血统229个样本中,检测到4645个表达HERV,其中4343个位于常染色体,852个有显著cis遗传调控;有15015个表达基因,5464个有cis调控
- 成人DLPFC中确实存在大量可检测HERV表达,而且相当一部分HERV表达能被附近遗传变异调控,为后面做rTWAS提供了前提
- 非洲组表达HERV数量和欧洲组差不多,但cis-heritable HERV数量更少,作者认为这可能部分来自样本量差异,也可能提示祖源差异
第3步:把SNP weights和精神疾病GWAS结果整合,做rTWAS
- Fig.1C:把表达预测模型和精神疾病GWAS summary statistics交叉整合,找出“被遗传风险牵引的表达特征”
-
TWAS:把表达预测模型和GWAS summary statistics整合
- rTWAS:把TWAS对象从普通gene扩展到HERV
- 输入:
- 每个gene/HERV的SNP weights
- 精神疾病GWAS summary statistics:只包含每个SNP的效应值、标准误、P值等,不需要个体级数据
- LD reference panel,即与建权重样本相匹配的CMC基因型数据:如果不控制LD,一个区域内多个SNP信号会重复计入
-
FUSION:用GWAS SNP效应和表达预测权重推断“遗传上预测的表达”是否与疾病风险相关
如果某个HERV附近的SNP既能预测HERV表达,又在GWAS中影响疾病风险,那么该HERV就可能出现rTWAS显著
- TWAS Z-score方向:正值通常表示遗传风险相关等位基因预测更高表达,负值表示预测更低表达,不等同于病例脑组织真实表达升降
- Bonferroni校正:严格多重检验校正,用于控制全局假阳性
-
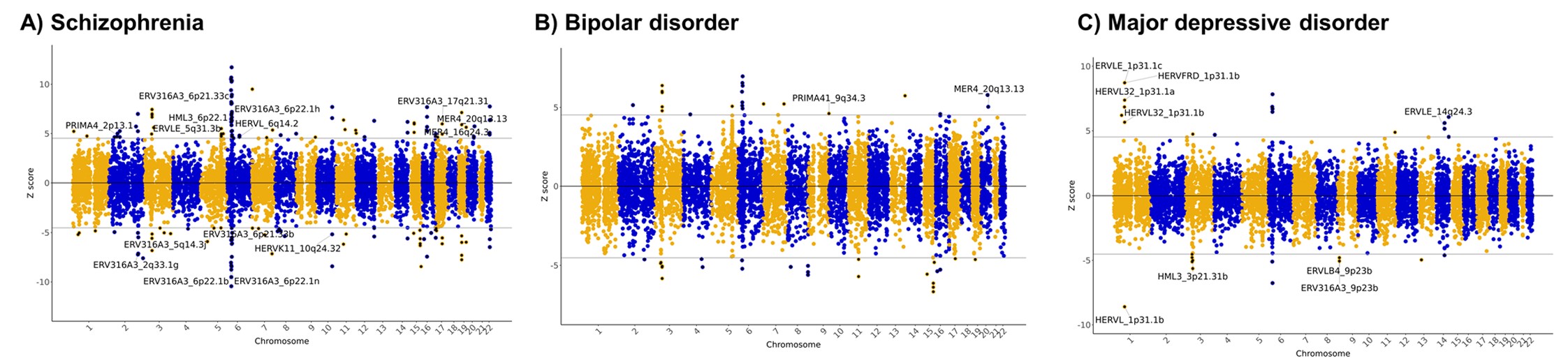
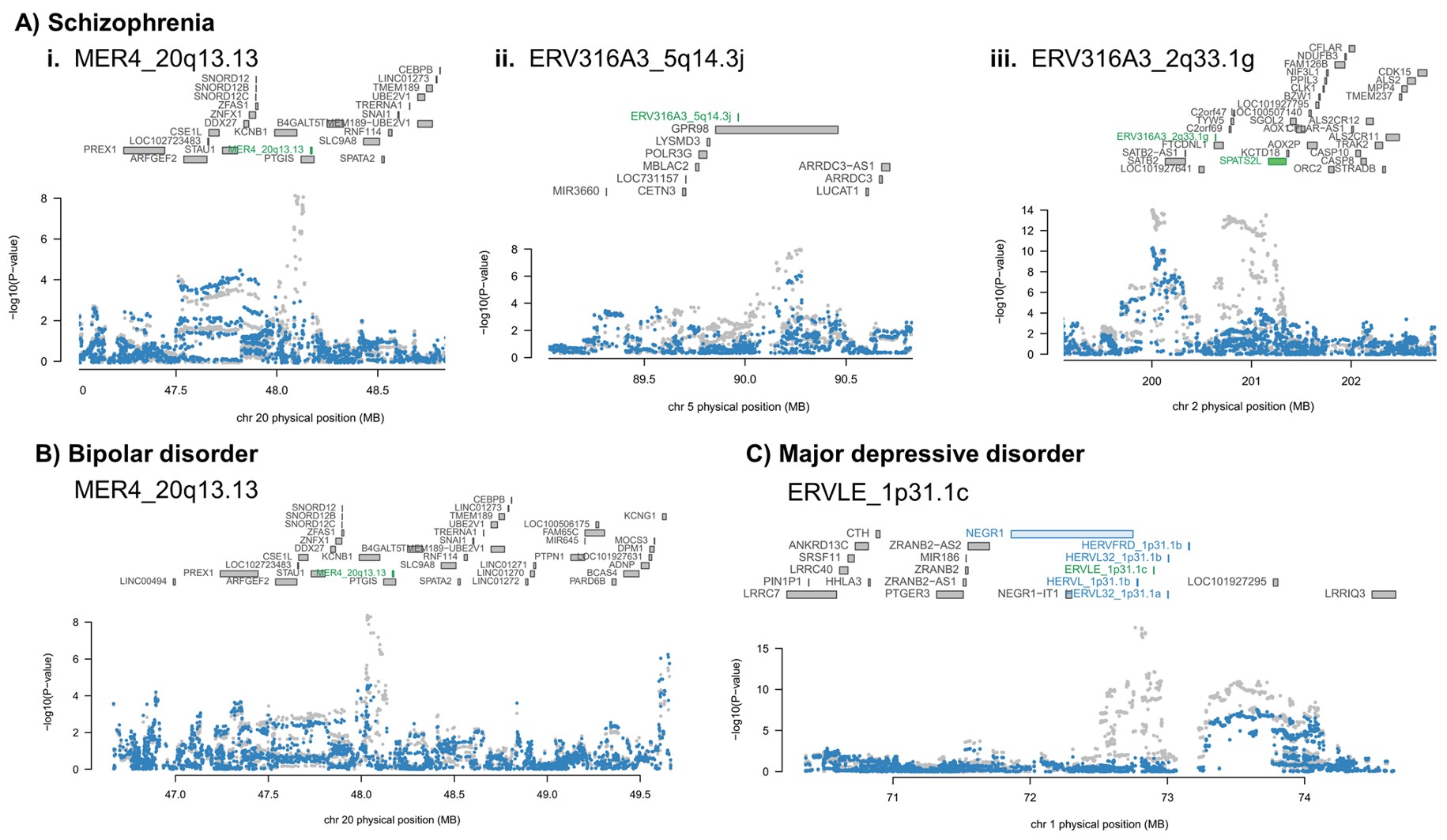
Manhattan biplot(曼哈顿图):分别对应精神分裂症、双相情感障碍和重度抑郁障碍。横轴是基因组位置,纵轴是TWAS Z-score(如果是
-log10(P)就是Manhattan plot,因为TWAS Z-score有正负,所以是bi);灰色水平线是Bonferroni显著性阈值;蓝色/黄色只是染色体分隔辅助色。这里找的特征同时包括基因和hERV,图上标注的是其中的hERV位点部分- MER4_20q13.13也在双相情感障碍和精神分裂症rTWAS中出现,且方向一致,提示MER4_20q13.13可能对应精神分裂症和双相障碍的共同遗传风险机制
- 抑郁症中HERV占比看起来较高,但后续真正进入高可信名单的主要是ERVLE_1p31.1c
- ADHD和自闭症谱系障碍没有显著HERV信号
- 这里的“风险表达特征”不是说病例中真实表达一定升高/降低,而是说遗传上预测的表达水平与疾病GWAS风险相关
- 这里出现的HERV信号需要后续条件分析判断它是不是独立于周围基因
第4步:用条件分析和fine-mapping筛出更可信的HERV
- Fig.1D/E:先看某个HERV信号是否独立于邻近gene,再看它在同一LD区域中是否最可能解释GWAS信号。最后得到high confidence risk HERVs
- 连锁不平衡(linkage disequilibrium, LD):附近SNP常一起遗传,所以一个GWAS峰可能牵连多个gene/HERV表达信号
- 后验纳入概率(posterior inclusion probability, PIP):PIP>0.5表示该表达特征比同一区域其它特征更可能解释关联信号
-
条件分析(conditional analyses):把同一区域的其它显著表达特征纳入后,看目标HERV是否还能解释GWAS信号
- 一个GWAS locus里可能有多个gene和HERV都显著,因为它们的预测表达模型共享同一批LD相关SNP。条件分析会把同一区域的显著表达特征放在一起,估计谁还能在联合模型中保留信号
- 这个HERV显著,是因为它自己能解释GWAS信号,还是因为它和旁边某个gene的预测表达高度相关?
- 如果还能解释,它就更像是独立信号;如果不能,可能只是“蹭到”附近gene或LD结构
- 方法:还是用FUSION,它不是拿真实表达矩阵直接做条件分析,而是基于遗传预测表达
- 比如
HERV1预测表达 = 0.3×SNP1 + 0.1×SNP2 - 0.2×SNP3 GeneA预测表达 = 0.2×SNP1 + 0.4×SNP2 + 0.1×SNP4- FUSION用SNP weights和LD矩阵计算二者预测表达的相关性
corr(HERV1预测表达, GeneA预测表达),判断HERV1和GeneA是不是由同一批或高度相关的一批SNP预测出来的,如果相关性很高,那么它们的TWAS信号可能来自同一个遗传信号;如果相关性低,则更可能是独立信号 - 之后FUSION条件/联合分析会进一步做类似这样的模型
disease GWAS signal ~ predicted GeneA expression + predicted HERV1 expression + predicted GeneB expression,然后看每个feature在联合模型中的P值,也就是论文中说的joint P(普通rTWAS是每个feature单独看HERV1 ~ disease risk/GeneA ~ disease risk,也称marginal association边际关联) - 如果HERV1单独显著,联合模型里也显著,作者就认为它是一个conditionally independent association
- 比如
- 用FOCUS在每个LD block内做TWAS fine-mapping(精细定位):在一个LD block中比较多个候选表达特征,估计谁最可能是因果信号。条件分析回答“是否独立”,fine-mapping回答“在同一区域多个候选表达特征中,谁更可能是因果/主解释信号”
- PIP越接近1,说明该表达特征越可能是该区域的主解释信号
- 条件分析后的局部区域图:每个小图上半部分是基因组上下文,下半部分是GWAS变异关联信号。灰点是原始GWAS中每个SNP与疾病风险的边际关联P值,蓝点是在条件化该locus中联合显著表达特征之后 每个SNP剩余的GWAS关联P值
- 如果蓝点明显比灰点低,说明:原始SNP→疾病风险信号有一部分和gene/HERV预测表达信号重合
-
GWAS峰:Manhattan plot上纵轴是
-log10(p),p值越小点越高。如果在某个基因组区域里,一群相邻SNP都和疾病显著相关,在图上形成一个像山峰一样的区域- 为什么一个区域会有“一群SNP”同时显著:连锁不平衡(linkage disequilibrium, LD)现象——附近SNP常常一起遗传。真正影响疾病风险的可能只有其中一个变异,但它周围一串SNP因为和它一起遗传,也会表现出疾病关联
- 所以GWAS峰通常说明:这个区域有遗传风险信号,但不一定立刻知道到底是哪个SNP、哪个gene或哪个HERV在起作用
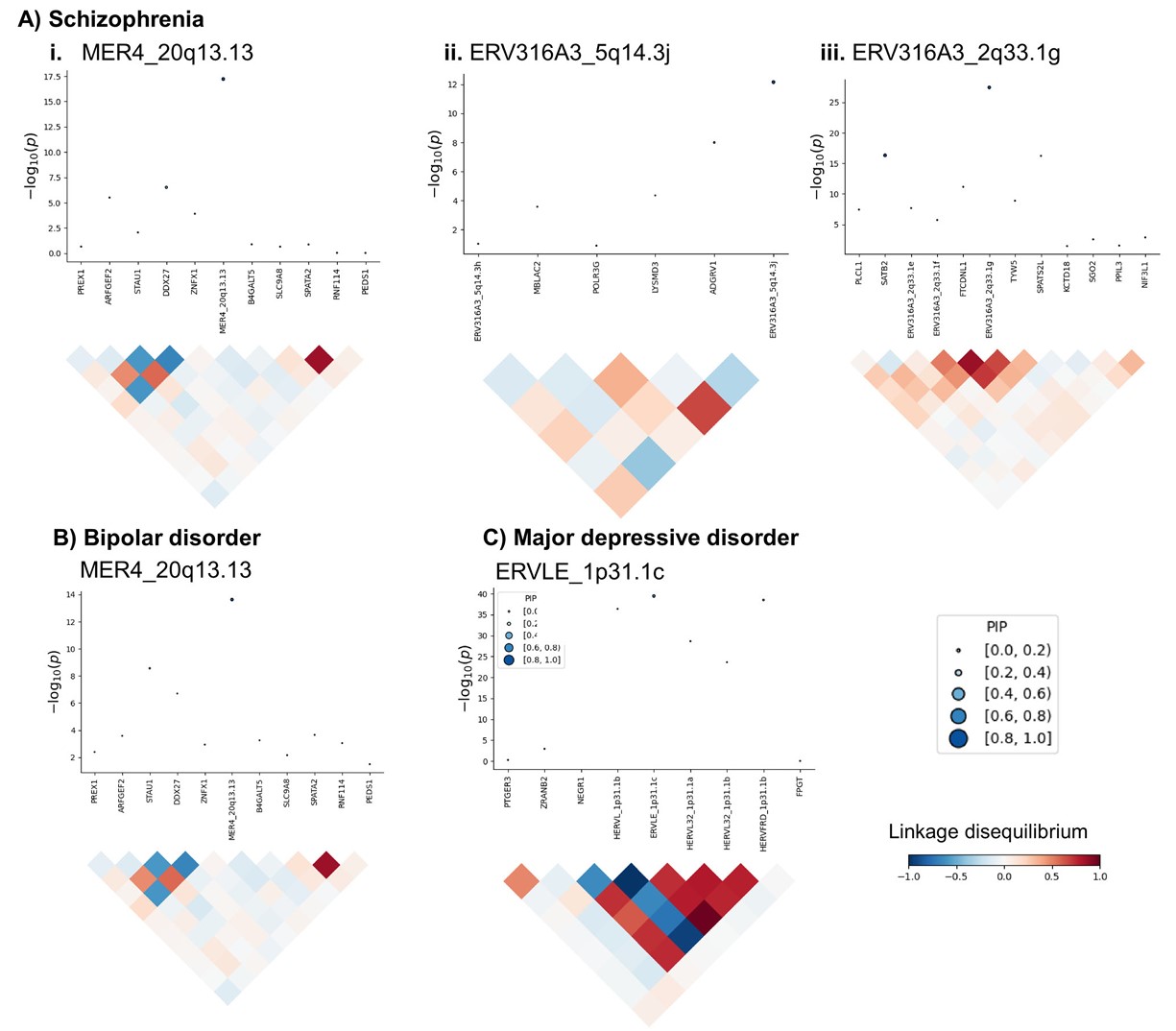
- 上半部分每个点是一个表达特征,纵轴是TWAS关联p值;点的大小和颜色代表PIP;下半部分是这些用遗传变异预测出来的表达特征的相关性
- 预测表达特征:比如说一个hERV位点和基因的SNP weights模型分别为
HERV1预测表达 = 0.3×SNP1 + 0.1×SNP2 - 0.2×SNP3和GeneA预测表达 = 0.2×SNP1 + 0.4×SNP2 + 0.1×SNP4,预测表达是根据一个人的基因型推算出来的表达倾向,预测表达特征就是这些SNP位点,表达特征的相关性就是“hERV1和geneA的遗传预测表达是不是由相似的一批SNP决定”,如果它们相关性很高,说明这两个表达特征的TWAS信号可能来自相同或相近的遗传信号 - 当然预测表达特征和LD有关:即使MER4和GeneA没有使用完全相同的SNP,但它们的SNP高度LD,那么它们的预测表达也会相关
- 因此,
预测表达相关性=SNP weights+SNP之间LD结构共同决定的相关性 - 如果两个feature间是正相关,就说明“预测HERV表达升高的遗传背景也倾向于预测GeneA表达升高”。以ERVLE_1p31.1c区域为例,它出现大片红/蓝结构,说明这个区域里很多候选表达特征的预测表达彼此相关或反相关,这意味着这个区域里多个gene/HERV的rTWAS信号可能互相牵连,不能只看谁的rTWAS P值最小;或者如果两个表达特征的预测表达高度相关,TWAS里它们可能都会显著,这时很难说到底是GeneA还是HERV1在解释GWAS信号,这些时候就要用FOCUS fine-mapping来判断谁更可能解释局部遗传信号
- 预测表达特征:比如说一个hERV位点和基因的SNP weights模型分别为
- 作者把“条件分析显著”且“PIP>0.5”的HERV定义为high confidence risk HERVs
GWAS → TWAS/rTWAS → 条件分析 → fine-mapping:一个逐层筛选、逐层提高可信度的流程
- GWAS:
SNP → 疾病风险,比如某个区域里很多SNP都和精神分裂症显著相关,于是形成一个GWAS峰,我知道“这个基因组区域和疾病风险有关”,但不知道它是通过哪个功能元件起作用的,可能是geneA/geneB/hERV/增强子,或者只是LD牵连- “GWAS发现的风险变异多数是非编码的,因此通常被认为可能影响局部基因调控”
- TWAS/rTWAS:
SNP → HERV/gene预测表达+SNP → 疾病风险,如果同一批SNP既能预测某个HERV/gene表达,又和疾病风险有关,TWAS就会说“这个HERV/gene的遗传预测表达与疾病风险相关”- 不是说“病人真实表达升高/降低”,而是“遗传上预测的表达高低与疾病风险相关”
- 条件分析:
- jointP和feature-level联合模型:与疾病风险相关的HERV/gene是它独立影响的,还是借用了邻近gene的影响力?或者说,HERV1在考虑GeneA/GeneB/GeneC之后,是否还显著?如果还显著,就是我们想要的真正有影响力的hERV位点
- Fig3条件化表达特征后GWAS SNP信号的下降:原始
SNP→疾病风险信号有一部分是gene/HERV预测表达信号影响的
- fine-mapping:在这个LD区域里,它是不是最可能解释该TWAS/GWAS信号的表达特征之一
- high confidence risk HERVs:
rTWAS显著+条件分析中独立+fine-mapping中PIP>0.5
第5步:敏感性分析和祖源外推
- 只用欧洲未患病对照构建TWAS weights,再与完整欧洲样本构建的weights比较
- 加入病例后,样本量增加133%
- 检测到cis-heritable HERV数量增加85%
- 用对照only权重和全样本权重做精神分裂症rTWAS,Z-score高度相关,Pearson r=0.95,但对照only分析显著结果减少16%
- 结论;结果不是明显由病例状态造成的偏差,把病例纳入建模不是为了做病例-对照差异,而是为了增加样本量、提高cis表达调控建模能力
- 跨祖源验证:
- 欧洲SNP weights+非欧洲GWAS:结果较弱,因为不同祖源的LD结构不同
- MER4_20q13.13在亚洲精神分裂症GWAS中有名义显著关联,但不能通过多重检验
- 非洲CMC weights+非洲裔GWAS:让weights和GWAS祖源匹配,结果没有发现Bonferroni显著表达信号。作者认为主要原因是非洲裔GWAS严重功效不足
- 结论;目前高可信结果主要限于欧洲血统,不能直接推广到其它祖源
- 欧洲SNP weights+非欧洲GWAS:结果较弱,因为不同祖源的LD结构不同
第6步:这些高可信HERV在基因组上是什么关系
- 这些HERV信号到底是独立HERV转录,还是普通gene转录本的一部分
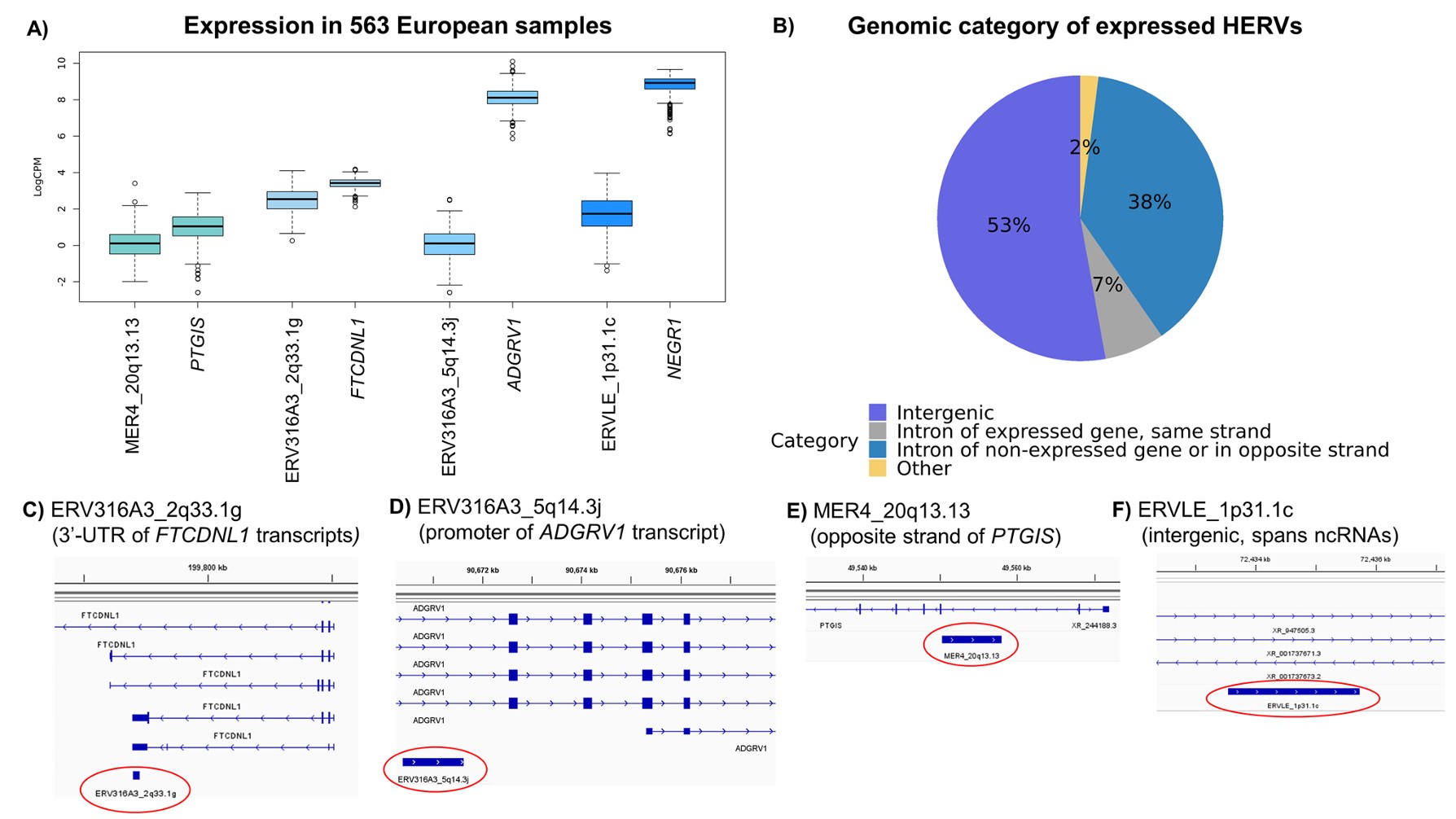
- A:比较高可信HERV和最近常规基因的logCPM表达,结果显示HERV表达普遍低于附近gene。作者认为这符合成人脑中HERV通常受表观遗传抑制的认识
- B:用HOMER注释HERV所在基因组区域,发现约98%的表达HERV位于基因间区或内含子区域
- intronic HERV:位于某个gene内含子中,不一定就是该gene转录本的一部分
- 如果HERV和gene同链且gene表达强,可能是gene isoform(基因mRNA前体剪接产生多种mRNA)的一部分;如果反义链、gene不表达或位于基因间区,更可能是独立非编码RNA
- C-D:ERV316A3_2q33.1g与FTCDNL1转录本3’UTR重叠;ERV316A3_5q14.3j位于ADGRV1转录本启动子区域。作者据此认为这两个rTWAS信号可能反映FTCDNL1和ADGRV1的特定isoform
- 这类HERV信号不能简单解释为“独立HERV RNA”,也可能是HERV参与形成的gene转录本结构。
- E-F:MER4_20q13.13位于PTGIS反义链;ERVLE_1p31.1c位于基因间区,最近基因是NEGR1,附近有ncRNA注释。作者认为这两个更可能代表新的非编码RNA
第7步:HERV可能参与哪些功能网络
- 在欧洲血统563个DLPFC样本中,把常规基因和HERV一起做WGCNA,得到16个表达模块,另有grey模块表示无法归类的特征
- 所有模块都包含HERV,说明HERV表达并不只集中在某个单一网络中,而是分布在多个表达程序里
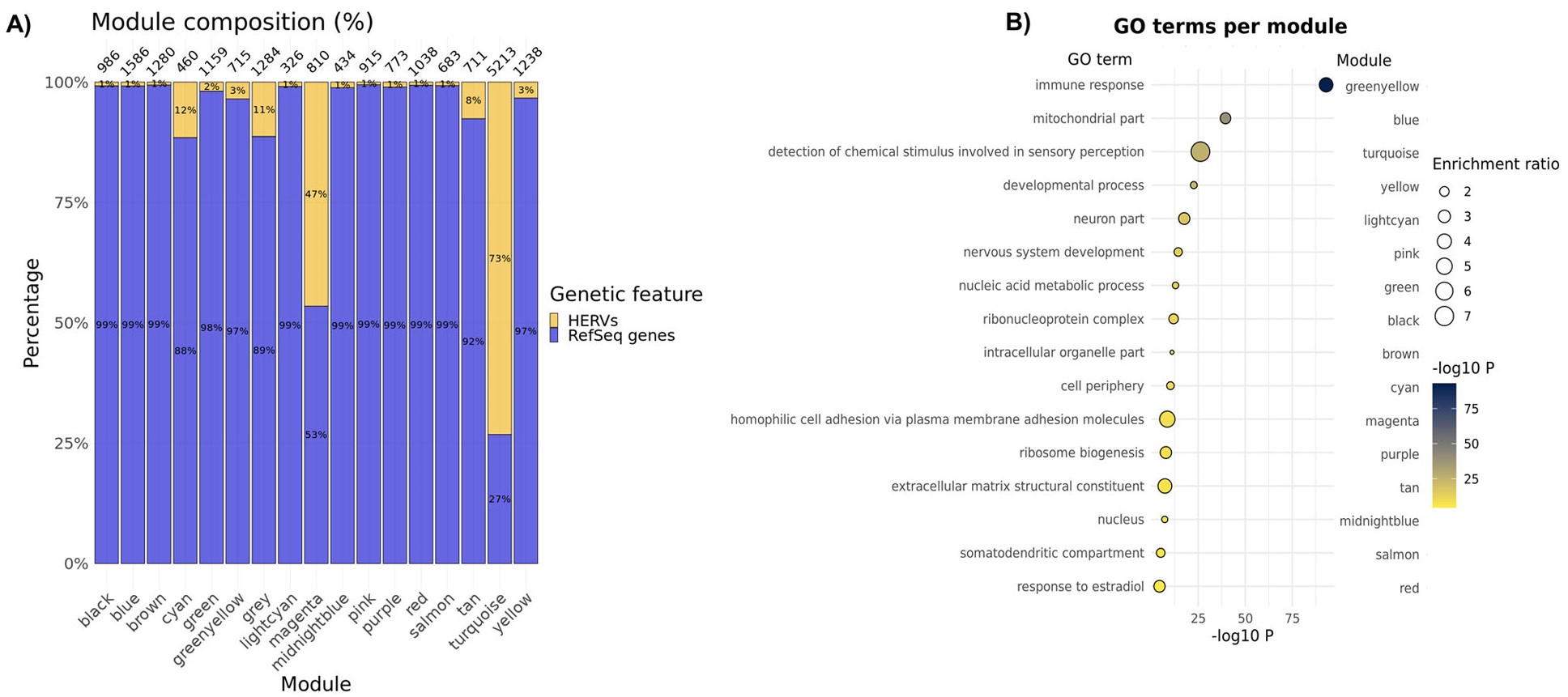
- A:每个共表达模块中gene和HERV的比例
- B:每个模块的GO富集结果
- cyan模块:synapse相关
- blue模块:mitochondria相关
- greenyellow模块:immune response相关
- turquoise模块:signal transduction相关
- 作者特别强调turquoise模块:包含1398个常规基因,占27%;包含3815个HERV,占73%;包括Table2中的所有高可信risk HERV;富集于信号转导相关GO term,如G protein-coupled receptor activity和detection of chemical stimulus
- 还做了两类稳健性检查:
- 调整样本来源、RIN、性别、病例状态、PMI、年龄、祖源PC和surrogate variables后,结果类似
- 在非洲血统229个样本中平行分析,turquoise模块也主要包含HERV,并且同样支持信号转导相关功能
- 结论:高可信HERV并不是孤立表达,而是落在一个HERV高度富集、并与信号转导相关gene共表达的大模块中
研究方法总结:遗传风险变异是否通过影响DLPFC中某些HERV位点的表达,从而参与精神疾病风险?
- RNA-seq定量gene和HERV表达
- 基因型数据建立表达预测模型(SNP weights),筛出能被cis遗传变异预测的HERV
- 把这些预测模型和精神疾病GWAS整合,做rTWAS,看看哪些HERV/gene的遗传预测表达与疾病风险相关(SNP~HERV/gene预测表达 + SNP~疾病风险)
- 用条件分析排除邻近gene牵连,看看哪些hERV位点在考虑邻近基因后还有影响力
- 用fine-mapping进一步判断哪个候选HERV是否最可能解释局部信号
- 用基因组上下文判断HERV是gene isoform的一部分还是独立ncRNA
- 用WGCNA从HERV和常规基因的共表达模块推断功能背景(GO富集)
为什么要分别分析欧洲和非洲:
- 欧洲和非洲血统是CMC中最大的、相对同质的两个子集:“同质”主要是遗传祖源上的同质,因为rTWAS需要用附近SNP预测表达,如果样本中混合很多祖源,SNP频率和LD结构会变复杂,预测模型会不稳定
- TWAS/rTWAS需要祖源匹配的SNP weights、LD reference和GWAS结果
- LD结构不同:某些SNP在欧洲人群中高度连锁,在非洲人群中可能不连锁
- 如果用欧洲weights去解释非洲GWAS,统计结果可能偏差
- 欧洲GWAS统计功效最高(欧洲精神疾病GWAS样本量更大、结果更稳定),所以用欧洲分析作为主分析
- 非洲样本用于两个目的:看HERV调控是否有祖源差异,以及测试结果能否外推
- 在表达结果中,非洲血统样本表达HERV数量与欧洲样本接近,但cis遗传调控的HERV重叠并不完全(非洲样本中852个cis-heritable HERV里,只有534个也在欧洲样本中显示cis调控)。虽然需要谨慎解释,因为两组样本量不同,但这提示可能存在祖源相关的HERV表达调控差异
- 测试欧洲样本中发现的高可信HERV是否能推广到其它祖源:跨祖源使用欧洲weights并不理想,因为不同人群LD结构不同
主要结论:
- 在DLPFC中检测到大量HERV表达,并且一部分受cis遗传调控
- 在欧洲血统563个样本中,作者检测到4594个表达HERV,其中4289个位于常染色体,1238个有cis-heritable expression
- 在非洲血统229个样本中,检测到4645个表达HERV,其中852个有cis-heritable expression
- 两组中表达的canonical genes数量接近,但欧洲组中检测到更多cis调控表达特征,可能与样本量更大有关
- rTWAS发现多个精神疾病相关的HERV表达信号
- 精神分裂症中发现163个Bonferroni显著表达特征,其中15个是HERV
- 双相情感障碍中发现47个显著表达特征,其中2个是HERV
- 重度抑郁障碍中发现29个显著表达特征,其中9个是HERV
- ADHD和自闭症谱系障碍中没有发现HERV显著信号
- 条件分析筛出独立于邻近gene的HERV信号
- 精神分裂症中有91个条件独立表达关联,其中6个是HERV
- 双相障碍中有30个条件独立关联,其中2个是HERV
- 重度抑郁障碍中有12个条件独立关联,其中2个是HERV
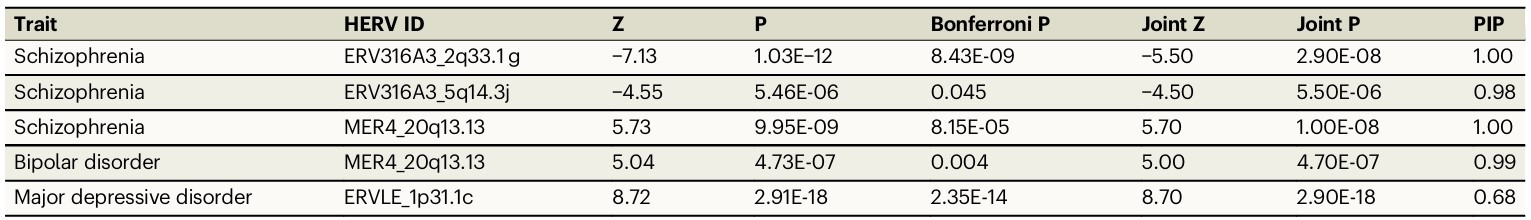
- fine-mapping进一步筛出高可信risk HERV
- 精神分裂症中有3个高可信HERV:ERV316A3_2q33.1g、ERV316A3_5q14.3j、MER4_20q13.13
- 双相障碍中MER4_20q13.13也是高可信HERV
- 重度抑郁障碍中ERVLE_1p31.1c是高可信HERV
- 把病例纳入权重构建并没有明显改变结果方向,主要作用是提高检测cis调控和rTWAS信号的统计功效:
- 加入病例使样本量增加133%,cis-heritable HERV检测增加85%
- 只用欧洲未患病对照做精神分裂症rTWAS时,结果与全样本weights高度相关,但显著信号减少16%
- 高可信HERV可能有不同分子含义
- HERV表达普遍低于附近gene
- 大多数表达HERV位于基因间区或内含子区
- ERV316A3_2q33.1g与FTCDNL1转录本3’UTR重叠,ERV316A3_5q14.3j位于ADGRV1转录本启动子区域,因此它们可能反映特定gene isoform
- 而MER4_20q13.13位于PTGIS反义链,ERVLE_1p31.1c位于基因间区,作者推测它们更可能是ncRNA
- 部分高可信HERV与预测增强子区域重叠,但需要实验验证
- 在UCSC Browser中查看发现ERVLE_1p31.1c、ERV316A3_2q33.1g和MER4_20q13.13所在位置包含ENCODE预测远端增强子,但ERV316A3_5q14.3j附近没有
- 由于缺少长读长RNA-seq来精确定义HERV转录本位置,增强子预测也需要实验验证
- 高可信risk HERV并不是孤立表达信号,而是落在一个HERV高度富集、与信号转导相关的共表达模块中
2024 NC–schERV+eQTL
样本情况:OneK1K PBMC单细胞数据GSE196830
- scRNA-seq数据+基因型数据:10X Genomics Chromium v2,来自981名健康供体,年龄范围20–90岁,总共约120万个单细胞
- syn50209110用于验证细胞类型特异HERV表达及其活性染色质富集
- Crohn’s disease(CD)部分用了患者单细胞数据GSE157477做表达验证
第1步:HERV GTF + gene GTF → merged GTF → CellRanger比对和计数 → 用基因注释细胞类型
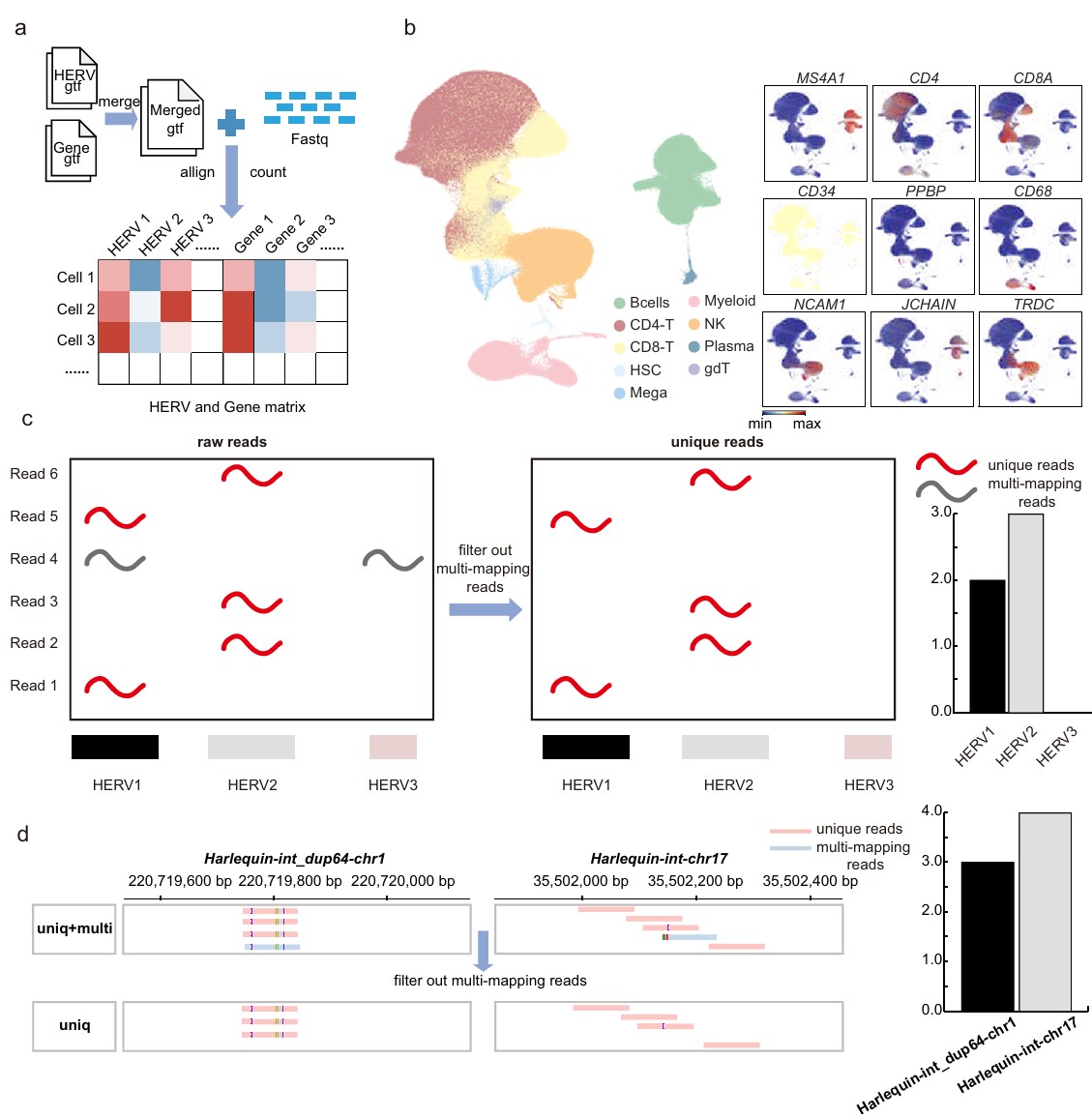
- 从UCSC Table Browser拿HERV注释(hg38),再和GENCODE v43蛋白编码基因注释合并
- 排除exonic overlap HERV:如果一个HERV和基因外显子重叠,单细胞读段可能来自宿主gene的成熟mRNA,而不是HERV自己启动的转录;作者为了保守,把这类HERV去掉,避免把宿主基因转录本中的片段误当成HERV独立表达
- CellRanger设置只保留唯一比对(unique mapping reads),图C和D就是举例说明只保留唯一比对后与全部读段数量的对比
- 过滤hERV位点:保留至少了20个细胞中表达的hERV
-
标准化:
(HERV计数×1e4)/(总计数)+log1p()转换- 单细胞的高变hERV筛选和差异表达用的是这个值,pseudobulk用的是counts相加
- 注释出9类免疫细胞:CD4-T、CD8-T、B、NK、myeloid、plasma、γδT、megakaryocytes和HSC,后续分析主要集中在CD4-T、CD8-T、NK、Bcells、Myeloid这5个主要细胞类型
第2步:确认PBMC中确实有大量HERV表达,并且多数不是简单跟随附近gene表达
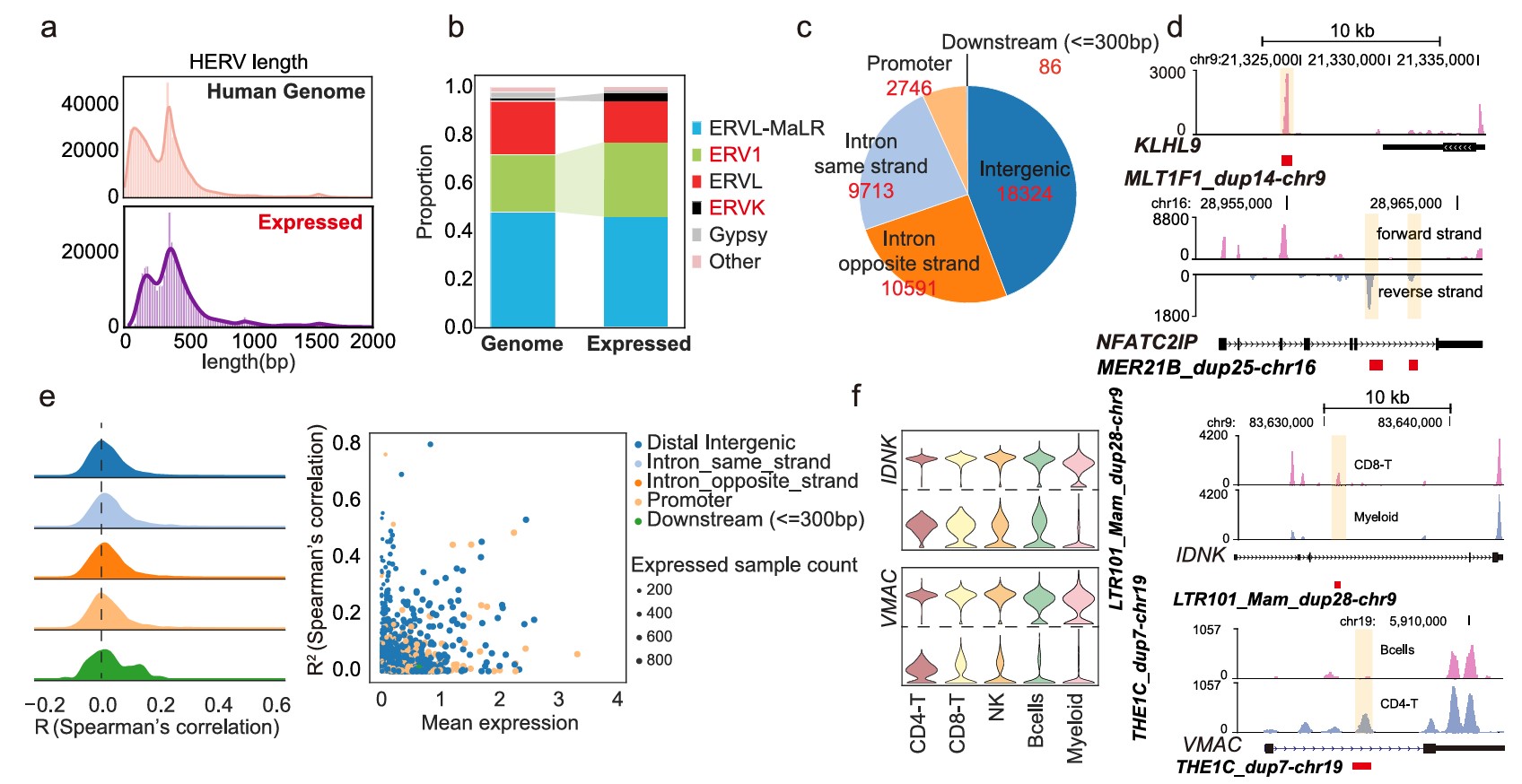
- 过滤hERV位点:保留在超过20个细胞中表达的位点,最终得到41460个表达HERV位点
-
A:比较了全基因组HERV和PBMC中表达HERV的长度分布。作者发现
PBMC中被检测到表达的HERV集合和人类基因组中所有注释HERV集合在长度组成上大体相似,并且很大一部分来自solo LTR或LTR相关片段- 如果表达HERV的长度分布和全基因组完全不同,比如只剩下特别短的HERV,或者只剩下特别长的HERV,那可能说明检测流程对某种长度的HERV有强烈偏好。现在作者看到二者整体相似,就可以说PBMC中检测到的HERV在长度层面没有明显偏离全基因组背景
-
B:表达HERV的家族分布。基因组中ERVL和ERV1占比较大,ERVK和ERV1表达比例更高
- MaLR是哺乳动物LTR逆转座子,ERVL-MaLR是和ERVL谱系关系很近的一类MaLR元素
-
C:表达HERV的基因组位置。表达的HERV并不只集中在基因附近,很多位于内含子或基因间区
- intron same strand:9713个,23%(内含子中HERV和附近gene在同一转录方向,可能更容易混入宿主gene转录信号)
- intron opposite strand:10591个,26%(反向,为同一个mRNA转录本的可能性较低)
- intergenic:18324个,44%(基因间区,更可能是独立转录或来自非编码调控区域)
- promoter/downstream较少
-
D:在基因组位置注释上展示读段在这个位置的堆积情况
- 如果很多RNA-seq reads都落在同一个HERV位点附近,图上就会形成一个明显的“峰”。这个峰说明:这个基因组位置确实有RNA转录信号
- MLT1F1_dup14-chr9对应的位置出现了一个清楚的局部峰,而且它位于KLHL9附近的基因间区,不是某个已知gene的外显子区域。作者因此认为它更像是一个真实转录信号,而不是随机噪声
-
E:每个HERV和最近gene之间的Spearman相关性。大多数HERV与附近gene相关性低,说明HERV表达并不只是附近gene表达的副产物
- 如果HERV在基因内含子里被数到,它是不是只是gene pre-mRNA的一部分?作者用相关性说明大多数HERV并没有简单跟着邻近gene一起变化
-
F:具体例子展示HERV和宿主gene表达模式不同。IDNK、VMAC这两个gene在多个细胞类型中比较稳定表达,但它们内含子附近的HERV,如LTR101_Mam_dup28-chr9和THE1C_dup7-chr19,在不同细胞类型中的表达变化更明显
- 左面小提琴图:虚线上是基因,下是位点
- 右面:不同细胞类型中峰不同
第3步:识别细胞类型特异HERV,并证明它们富集在细胞类型特异活性染色质区域
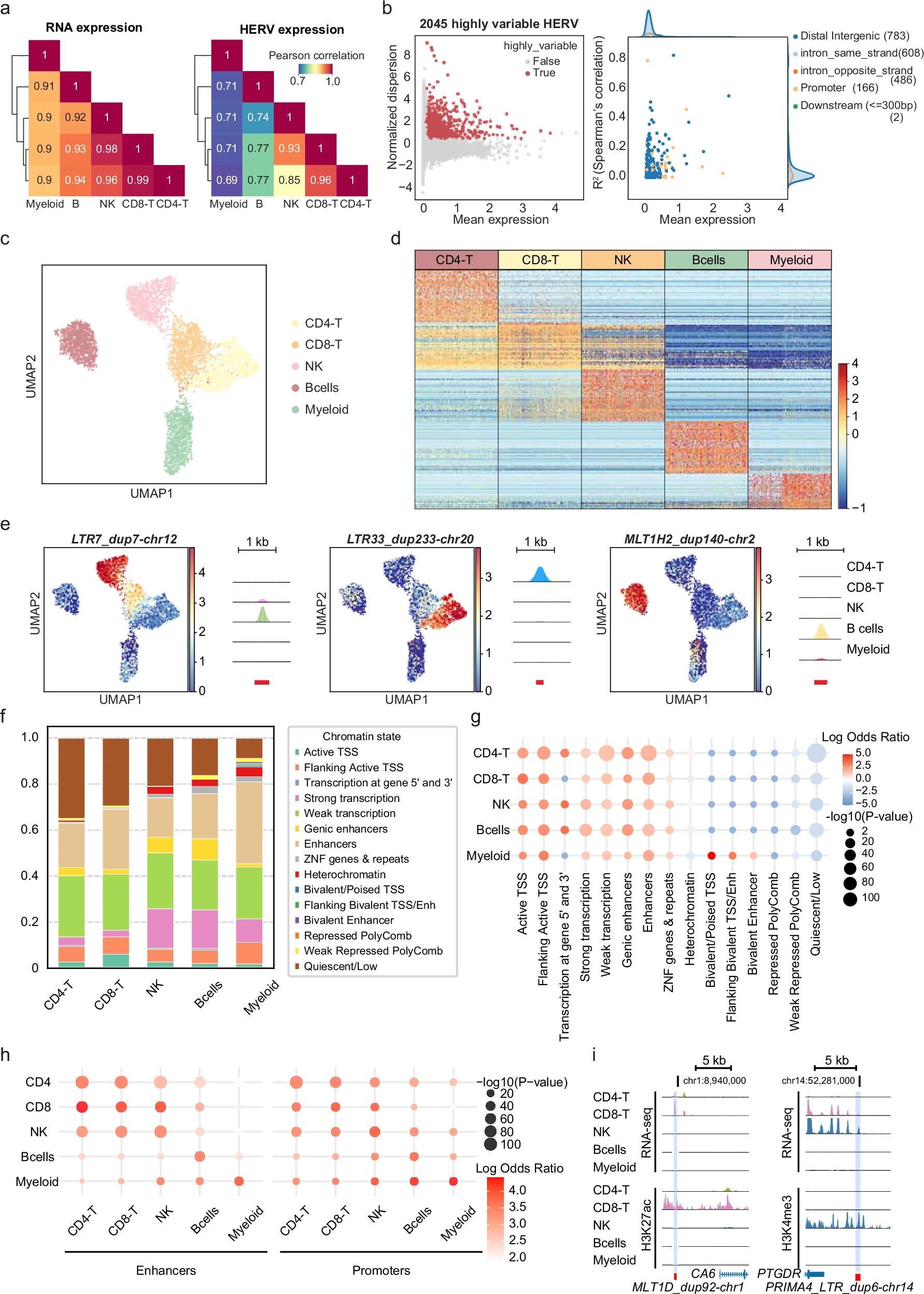
- A:gene表达和HERV表达的细胞类型相似性比较,左边是gene表达在5类主要免疫细胞之间的相关性,右边是HERV表达的相关性。结果是HERV表达谱在不同细胞类型之间相似性更低,说明HERV表达比普通gene更能体现细胞类型差异
- B:根据平均表达和离散度筛选高度变异HERV,右图是高变HERV与相关基因的Spearman相关系数散点图,显示这些高度变异HERV与邻近gene仍然相关性低
-
C:看这些已知细胞类型在只有高变HERV的UMAP中能不能分开——作者已经知道哪些细胞是CD4-T、CD8-T、NK、B、Myeloid,然后把这些细胞放到“只由HERV表达决定”的低维空间里,如果同一种细胞类型仍然聚在一起,就说明HERV表达本身携带细胞类型信息。结果显示只用高变HERV做UMAP,五类主要免疫细胞仍然能在低维空间里分开
- 需要注意的是,这里不是用hERV划分细胞类型,只是看各细胞类型的高变hERV表达情况是否相同
- 比较特别的地方:图里每个点不是一个细胞,而是一个
某个供体 × 某个细胞类型(用pseudobulk聚合每个供体的每个细胞类型的细胞)。可能是因为HERV表达太稀疏,作为主成分的hERV位点在部分细胞中表达可能为0;也可能是与后续eQTL的输入形式统一,都用pseudobulk
-
D:细胞类型特异HERV表达热图,用Scanpy的
sc.rank_genes_groups()识别细胞类型特异HERV,判定标准是Bonferroni校正后P值<1e-5且log2FC>2,最终得到1936个细胞类型特异HERV -
E:展示几个细胞类型特异HERV例子
- LTR7_dup7-chr12:主要在NK细胞表达
- LTR33_dup233-chr20:主要在CD4-T细胞表达
- MLT1H2_dup140-chr2:主要在B细胞表达
-
F:把细胞类型特异HERV映射到Roadmap Epigenomics的15种染色质状态,结果显示细胞类型特异HERV主要落在活性染色质状态
- Roadmap Epigenomics把基因组分成15类染色质状态,例如Active TSS(活性转录起始位点)、Enhancers(增强子)、Strong transcription(强转录区域)、Repressed PolyComb(Polycomb抑制区域,一类进化上高度保守的转录抑制因子)、Quiescent/Low(低活性区域)
- 如果HERV富集在Active TSS或Enhancers,说明它可能受到细胞类型特异表观调控,甚至可能本身具有启动子/增强子功能
- G:用Fisher精确检验比较细胞类型特异HERV和背景HERV在不同染色质状态(Roadmap Epigenomics)中的重叠比例,证明细胞类型特异HERV显著富集于活性染色质(相当于用统计富集检验证明F的结果)
-
H:沿用G的统计方法,看细胞类型特异HERV是否富集在对应细胞类型的enhancer/promoter区域中。结果显示,某一类细胞中特异表达的HERV,更容易落在该细胞类型自己的enhancer或promoter区域
- enhancer或promoter由ENCODE的H3K27ac/H3K4me3 ChIP-seq定义
-
I:具体例子展示HERV和细胞类型特异表观信号重叠。
- MLT1D_dup92-chr1位于CD8-T细胞enhancer,并主要在CD8-T中表达
- PRIMA4_LTR_dup3-chr14位于NK细胞promoter区域,并主要在NK中表达
- 还是ChIP-seq数据
- H3K27ac ChIP-seq track:显示该区域是否有活跃enhancer/promoter信号
- H3K4me3 ChIP-seq track:显示该区域是否有活跃promoter/TSS(转录起始点)信号
- DNase-seq:检测开放染色质区域,显示该区域是否更容易被转录因子结合
第4步:做HERV cis-eQTL,寻找调控HERV表达的遗传变异
-
表达定量性状基因位点(expression quantitative trait locus, eQTL):如果某个SNP的不同基因型对应某个HERV表达高低不同,就说明这个SNP可能调控该HERV表达
- cis-eQTL:只看HERV附近的SNP,本文范围是HERV TSS上下游1Mb
- eHERV:被SNP调控表达的HERV
- eSNP:调控HERV表达的SNP
-
conditionally independent eQTL:同一个HERV附近可能有很多SNP显著,但它们之间可能有LD,也就是一起遗传。TensorQTL的
cis.map_independent()会尝试在同一区域中筛出相对独立的调控信号,避免把同一遗传信号重复计数
- 用TensorQTL做线性模型,分别在不同细胞类型中检测SNP-HERV表达关联
- 作者不是拿单个细胞直接做eQTL,而是用pseudobulk的方法:eQTL的表型不是单个细胞表达,而是每个供体在某个细胞类型中的pseudobulk HERV表达
- 控制协变量:
- 性别、年龄
- 6个基因型PC:控制群体遗传结构,避免不同祖源背景造成假关联
- 2个PEER因子:从表达矩阵中估计隐藏混杂因素,如批次、样本处理差异等
- 控制协变量:
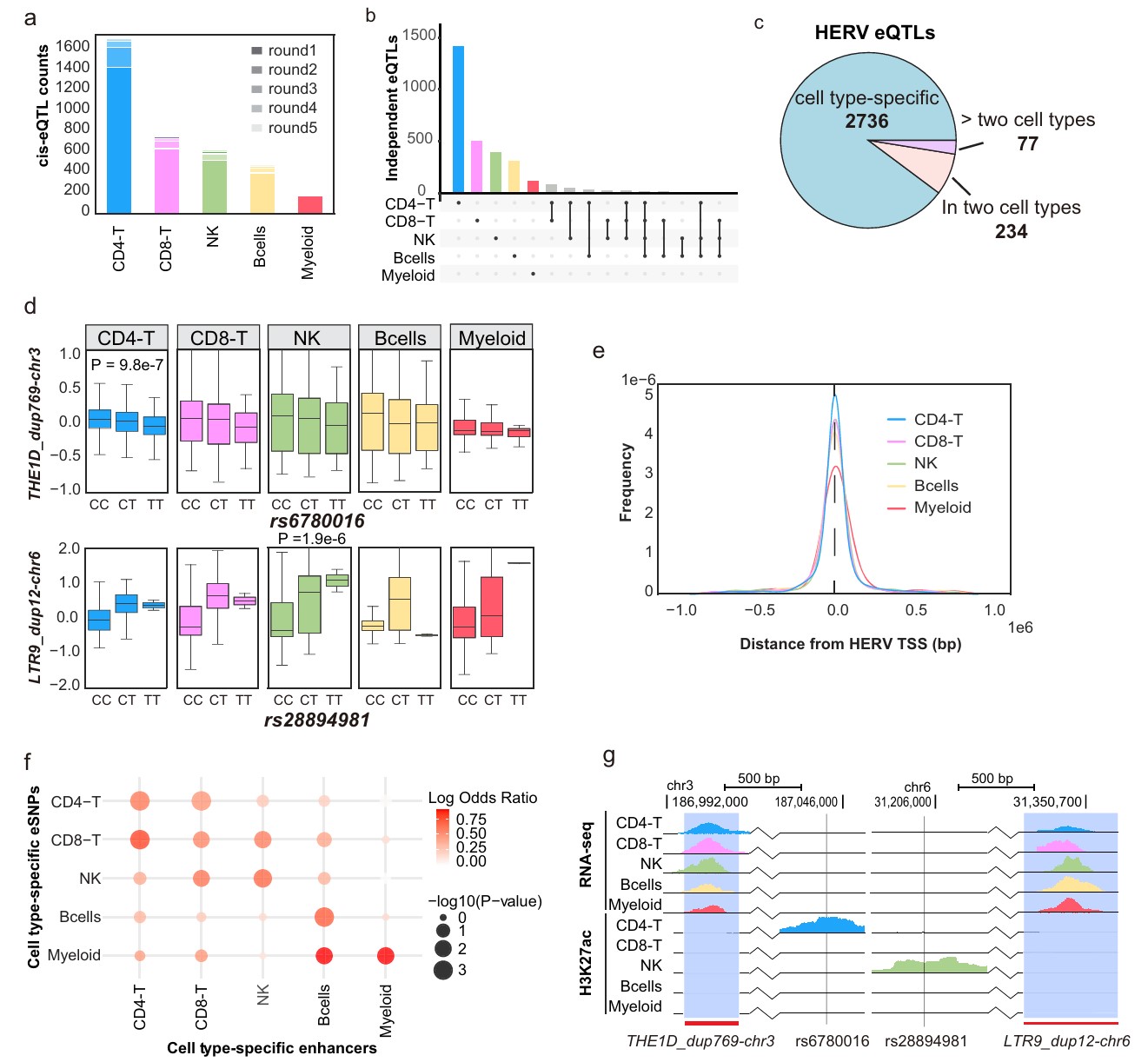
-
A:五类免疫细胞中识别到的独立HERV eQTL数量
- 图例里的round1/round2/round3/round4/round5指的是TensorQTL在识别条件独立cis-eQTL时的迭代轮次。对每个HERV,在其TSS上下游1Mb内可能有很多SNP与它的表达相关。但这些SNP之间往往有LD,很多显著SNP其实代表同一个遗传信号
- round1可以理解为:对每个HERV先找最强的主eQTL信号,也就是最显著的lead eSNP-HERV关联
- 如果某个新的SNP在控制round1 lead SNP后仍然显著,它就可能代表第二个独立调控信号,也就是round2
- 如果某个细胞类型中round1占绝大多数,说明大部分eHERV只有一个主要cis调控信号;如果出现round2/3/4/5,说明部分HERV附近存在多个相互独立的调控变异
- CD4-T最多,Myeloid最少
- 共3463个条件独立cis-eQTL,涉及1805个HERV和2888个SNP
- 图例里的round1/round2/round3/round4/round5指的是TensorQTL在识别条件独立cis-eQTL时的迭代轮次。对每个HERV,在其TSS上下游1Mb内可能有很多SNP与它的表达相关。但这些SNP之间往往有LD,很多显著SNP其实代表同一个遗传信号
-
B/C:Upset plot/饼图,展示不同细胞类型之间eQTL共享情况
- 显示2736个eQTL只在单一细胞类型中出现,234个出现在两个细胞类型,77个出现在超过两个细胞类型大多数HERV eQTL是细胞类型特异的
- 说明HERV表达的遗传调控大多不是全PBMC共享的,而是发生在特定细胞类型中
-
D:具体eQTL例子,横轴是基因型CC/CT/TT,纵轴是HERV表达
- THE1D_dup769-chr3与rs6780016:只在CD4-T中显著
- LTR9_dup12-chr6与rs28894981:只在NK中显著
- 只有特定细胞类型中,不同基因型之间的表达差异才明显
- 细胞类型特异eQTL不一定是因为该HERV只在某类细胞达,多数cell type-specific eHERV其实在多个细胞类型都表达。差别在于“遗传变异对它表达的调控作用”只在某些细胞类型显现
- E:eSNP到目标HERV TSS的距离分布,多数eSNP集中在HERV TSS附近约200kb内,符合cis调控特征——调控变异通常离目标feature不太远
-
F:eSNP与细胞类型特异enhancer的统计富集分析,行是cell type-specific eSNP,列是cell type-specific enhancers。如果CD4-T特异eSNP更常落在CD4-T enhancer,说明这种遗传调控依赖CD4-T特异染色质环境,结果显示eSNP富集在对应细胞类型enhancer
- 把遗传调控和表观调控连起来:SNP之所以只在某类细胞调控HERV,可能因为该SNP所在区域只在这类细胞中是活性enhancer
-
G:展示两个区域的RNA-seq和H3K27ac轨迹。HERV表达不仅有细胞类型特异性,而且其遗传调控也具有细胞类型特异性,这种特异性很可能由细胞类型特异enhancer状态决定——只有当调控SNP所在区域在某个细胞类型中成为活跃enhancer时,这个SNP-HERV eQTL才会在该细胞类型中显现
- RNA-seq轨迹:这个HERV位点在不同细胞类型中有没有转录信号。可以看到THE1D_dup769-chr3不是只在CD4-T表达,而是在五类细胞里都有表达。排除“因为THE1D_dup769-chr3只在CD4-T表达,所以只能在CD4-T检测到eQTL”的解释
- H3K27ac轨迹:eSNP附近是不是处在细胞类型特异的活跃增强子区域。rs6780016附近在CD4-T细胞中有明显H3K27ac峰,而其它细胞类型不明显,说明rs6780016所在区域在CD4-T中像一个活跃enhancer
- 这张图与D图对应:D显示rs6780016和THE1D_dup769-chr3表达的关联只在CD4-T中显著,这张图从具体表达层面验证D图结果
- rs6780016位于CD4-T特异enhancer,调控THE1D_dup769-chr3
- rs28894981位于NK相关活性区域,调控LTR9_dup12-chr6
第5步:把HERV eQTL和免疫相关疾病GWAS整合,发现HERV与自身免疫病显著相关
-
基于汇总数据的孟德尔随机化(summary-data-based Mendelian randomization, SMR):用GWAS summary statistics和eQTL summary statistics,测试一个表达特征是否和疾病风险共享遗传信号
-
SNP → HERV表达变化+SNP → 疾病风险变化:如果两者方向和效应一致,就提示HERV表达可能参与该SNP到疾病风险的通路 - HEIDI检验:用来排除“两个不同因果变异只是因为LD靠得近而看起来有关”的情况。如果pHEIDI太低,说明周围SNP效应模式不一致,可能不是同一个因果变异
- 本文把q-value<0.05且pHEIDI>0.01作为显著结果
- 作者做了三类SMR:HERV-gene、HERV-disease、gene-disease
-
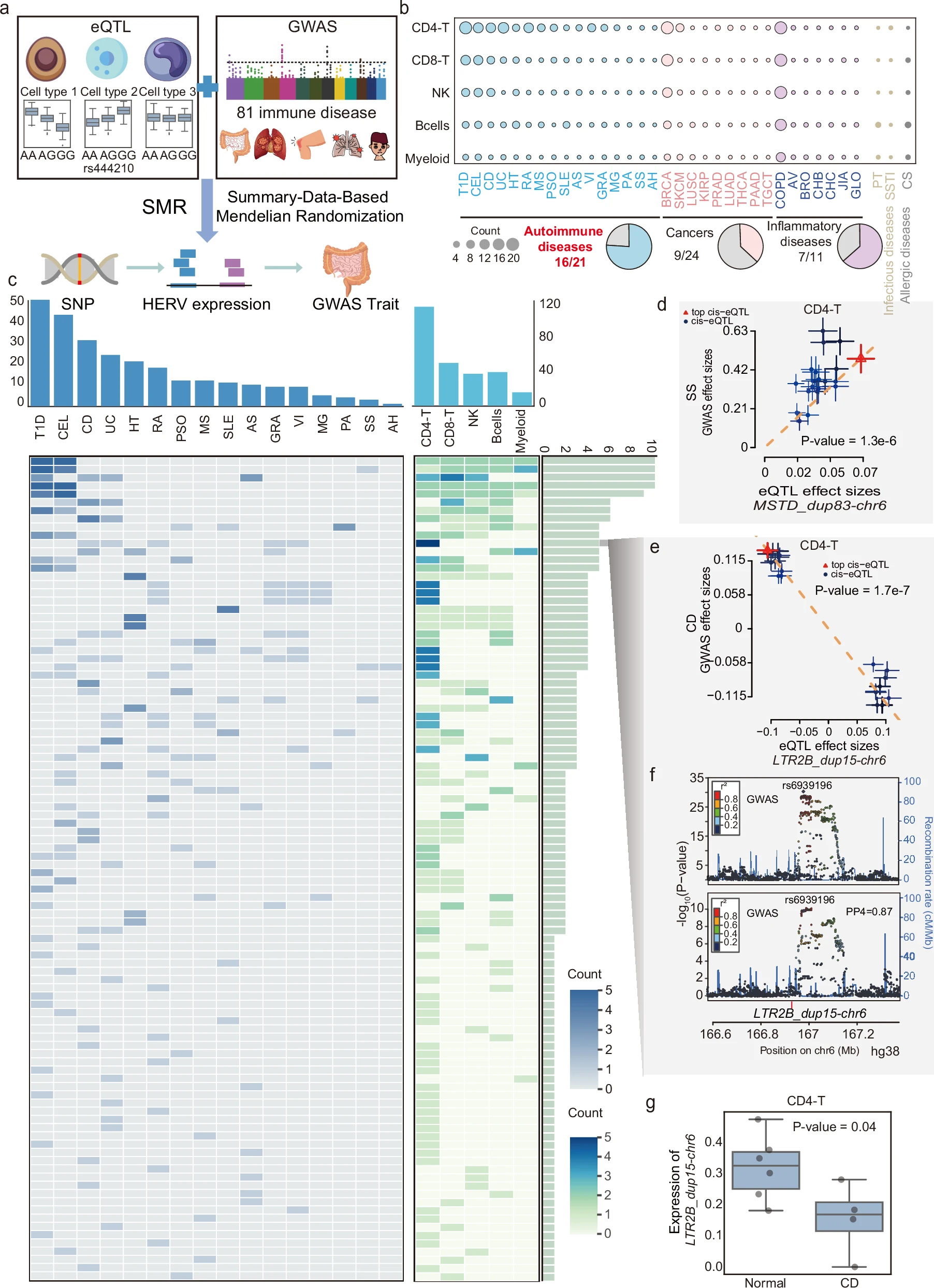
-
A:SMR分析示意图。
细胞类型特异HERV eQTL+81种免疫疾病GWAS→SMR→SNP-HERV表达-疾病关联 -
B:展示不同细胞类型中HERV与各类疾病的显著关联数,以及各疾病类别中有多少疾病能检测到HERV关联
- 548个显著HERV-disease关联,涉及196个eHERV和35种疾病
- 其中自身免疫病最突出,21种自身免疫病中有16种和100个细胞类型特异HERV有关
-
C:按疾病和细胞类型统计显著关联数量,并展示不同HERV和不同自身免疫病、不同细胞类型的关系
- HERV关联最明显的是T1D、CEL、CD、UC等消化系统或肠道免疫相关疾病
-
D:以MSTD_dup83-chr6为例,展示其eQTL效应和Sjogren’s syndrome GWAS效应的关系。每个点是一个SNP,横轴是该SNP对HERV表达的效应,纵轴是该SNP对疾病风险的效应,误差条是SNP效应的标准误差,虚线表示SMR估计的效应方向,红点通常表示top cis-eQTL。如果SNP对HERV表达的效应和对疾病的效应呈线性关系,说明二者可能共享遗传驱动
- GeneHancer(整合增强子-基因互作证据的数据库)记录该位点和7个HLA基因有互作,这和HLA在自身免疫病中的重要性一致
- E:展示LTR2B_dup15-chr6的eQTL效应和Crohn’s disease(CD) GWAS效应,LTR2B_dup15-chr6与Crohn’s disease在CD4-T中负相关。作者发现该HERV在CD4-T中和CD风险负相关(遗传上预测LTR2B_dup15-chr6表达更高时,CD风险更低)
-
F:LocusZoom图,上方是CD GWAS信号,下方是LTR2B_dup15-chr6在CD4-T细胞中的eQTL信号,每个点是一个SNP,横轴是chr6上的物理位置,纵轴是
-log10(P-value),颜色表示每个SNP与标注的lead SNP(rs6939196)之间的LD强度,通常用r²表示,越接近1则LD越强。图里峰越高,代表这个位置附近的SNP和对应性状的关联越强- 两个信号在rs6939196附近共定位,SNP.H4=0.87,支持LTR2B_dup15-chr6和CD共享因果SNP
-
共定位(colocalization):检测两个信号是否由同一个因果变异驱动。在图中通过看GWAS峰和eQTL峰是否重叠来判断——
最高峰位置接近+lead SNP相同或高度LD+周围SNP的显著性峰形相似,这张图里两个信号都集中在rs6939196附近 - H4 posterior probability:两个性状共享同一因果变异的后验概率。这里SNP.H4=0.87,与共定位结果一起说明“有较强证据支持CD GWAS信号和LTR2B_dup15-chr6 eQTL信号共享同一个因果SNP”
- 为什么要在E的基础上画这个图:排除SMR中可能遇到“SNP A真正调控HERV表达、SNP B真正影响CD风险、SNP A和SNP B因为LD很强,所以看起来像同一个信号”的问题,如果两个峰错开就说明是两个不同信号
- G:使用GSE157477的CD患者和健康供体单细胞数据,比较LTR2B_dup15-chr6在CD4-T中的表达,这是用患者数据证明LTR2B_dup15-chr6在CD4-T细胞中与CD状态相关,且在CD患者中表达降低
第6步:进一步连接“HERV→gene→disease”,提出LTR2B_dup15-chr6可能调控RNASET2影响Crohn’s disease
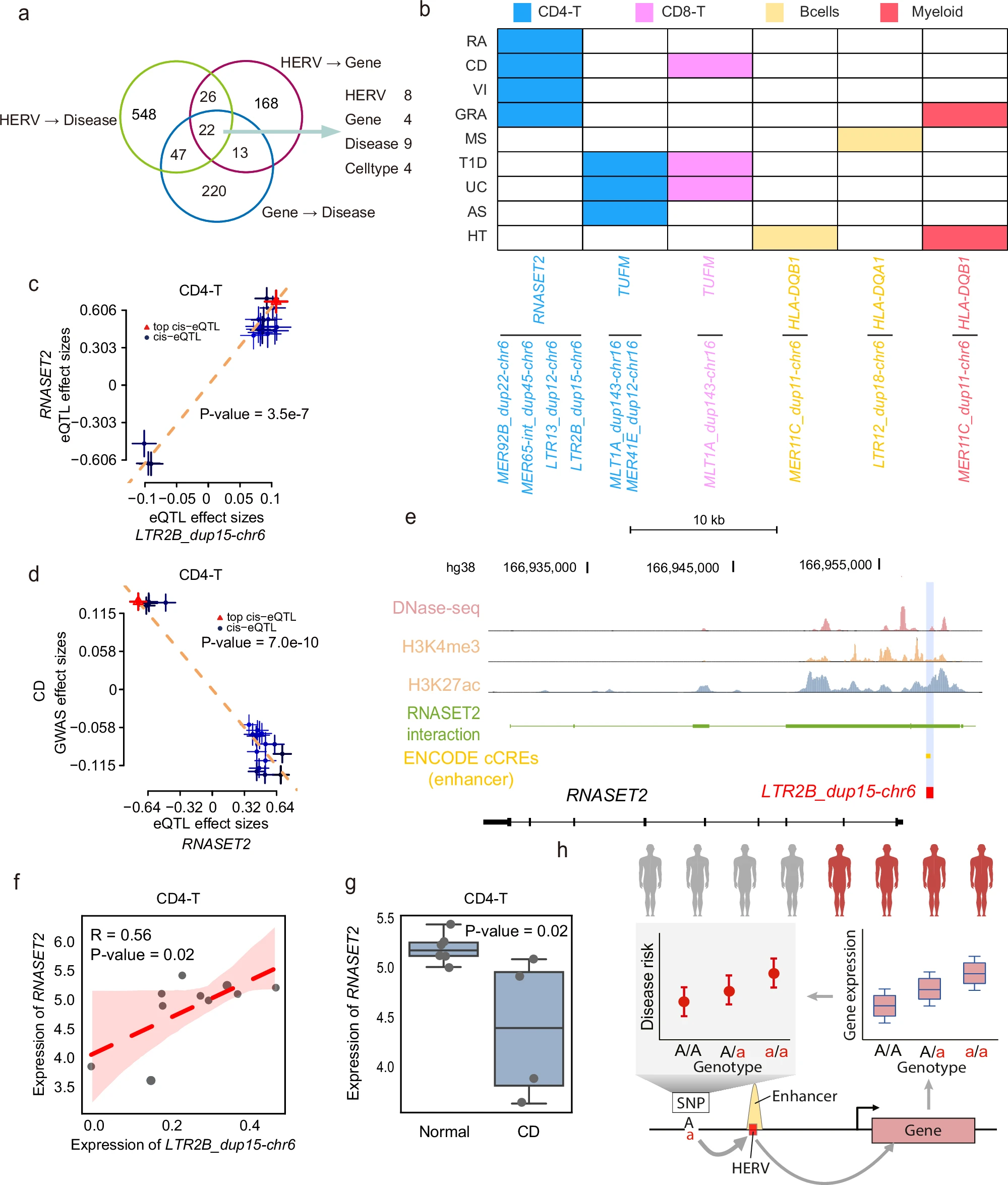
-
A:HERV→Disease、HERV→Gene、Gene→Disease的Venn图。如果某个HERV和某个gene表达相关,而这个HERV和gene又都和同一种疾病相关,那么它可能构成一个候选通路
- 共22个交集,涉及9种自身免疫疾病、4种细胞类型、8种HERV和4个基因
-
B:展示三类关联重叠后的具体“HERV-gene-disease-cell type”组合
- 重点例子是CD4-T里的LTR2B_dup15-chr6、RNASET2和Crohn’s disease
- C:展示LTR2B_dup15-chr6 eQTL效应和RNASET2 eQTL效应的关系,二者呈正相关,P=3.5e-7。结果显示LTR2B_dup15-chr6和RNASET2表达共享遗传调控信号,作者认为LTR2B_dup15-chr6可能调控RNASET2表达
- D:展示RNASET2 eQTL效应和CD GWAS效应的关系,二者呈负相关,P=7.0e-10,说明RNASET2表达也和CD风险有遗传关联,把HERV和gene都连接到了同一个疾病
-
E:整合CD4-T细胞中的DNase-seq、H3K4me3、H3K27ac、GeneHancer互作和ENCODE cCREs,LTR2B_dup15-chr6这个位置有明显增强子信号,并且位于RNASET2上游约1kb,GeneHancer还记录了该增强子和RNASET2之间的互作,说明LTR2B_dup15-chr6可能通过增强子样作用影响RNASET2表达
- 图下方黑色结构是RNASET2基因结构,红色小块是LTR2B_dup15-chr6
- RNASET2 interaction:来自GeneHancer,表示哪些基因组调控区域被记录为与RNASET2有调控联系
- ENCODE cCRE(candidate cis-regulatory elements):图中的黄色小块,表示ENCODE预测的候选顺式调控元件,其中这里标注为enhancer
-
怎么看出“LTR2B_dup15-chr6可能通过增强子样作用影响RNASET2表达”
- 空间位置接近
-
LTR2B区域有enhancer信号
- H3K27ac轨迹在LTR2B附近有明显峰,作者说“significant enhancer signals”主要就是依据这类组蛋白修饰和开放染色质信号
- DNase-seq在附近也有峰,说明这个区域染色质开放,具有被转录因子访问的潜力
- ENCODE cCREs(enhancer)轨迹在LTR2B附近有黄色enhancer标注,说明ENCODE把这个区域预测为候选增强子
- RNASET2 interaction显示:绿色interaction区域延伸到LTR2B_dup15-chr6附近,说明这个含LTR2B的enhancer区域被GeneHancer记录为RNASET2相关调控元件
- 不过这里不能证明该hERV位点本身就是增强子核心元件或者直接导致RNASET2表达变化
- F:GSE157477数据集的CD4-T细胞中LTR2B_dup15-chr6和RNASET2样本级表达散点图,显示二者正相关
- G:RNASET2在正常CD4-T中高于CD患者CD4-T,P=0.02
-
H:作者提出的机制模型——
遗传变异 → 影响HERV/enhancer活性 → 改变附近gene表达 → 改变疾病风险,具体到本文就是rs6939196附近遗传信号 → LTR2B_dup15-chr6表达/增强子活性 → RNASET2表达 → Crohn’s disease风险
这篇文章中的eQTL+SMR:把SNP作为媒介,讨论hERV-gene-疾病三者的相关性
-
HERV-eQTL summary statistics:每个细胞类型里,每个供体聚合成pseudobulk HERV表达,然后用TensorQTL检测
SNP基因型 → HERV表达,并且只看HERV TSS上下游1Mb内的cis变异,最后得到条件独立eQTL - 疾病GWAS summary statistics:作者收集了81种免疫相关疾病GWAS,包括21种自身免疫病、24种癌症、11种炎症疾病、6种过敏疾病和19种感染疾病
- public gene eQTL results:作者收集了欧洲人群公共gene eQTL结果
- 对于一个HERV或gene,SMR先找它的显著eQTL SNP,通常是top associated eQTL,作为工具变量SNP(显著影响HERV表达或gene表达,同时在GWAS里也有疾病效应估计)
-
HERV→Disease:
- 输入
HERV-eQTL(SNP → HERV表达)、Disease GWAS(SNP → 疾病风险) - 如果这个SNP显著影响HERV表达,同时它对疾病风险也有方向一致/可解释的效应,则认为这个HERV与疾病存在遗传层面的调控关联
- 输入
-
HERV→Gene:
- 输入
HERV-eQTL、gene eQTL(SNP → gene表达) - 如果同一个SNP既影响HERV表达,又影响某个gene表达,SMR显著,并且pHEIDI没有提示明显异质性,作者就认为这个HERV和gene之间存在遗传层面的调控关联
- 输入
-
Gene→Disease:
- 输入
gene eQTL、Disease GWAS - 如果某个gene的eQTL SNP也和某个疾病GWAS风险相关,SMR显著,就认为该gene表达和疾病存在遗传层面的调控关联
- 输入
- 最后提出
LTR2B_dup15-chr6-RNASET2-CD这条路径:- HERV→Disease:LTR2B_dup15-chr6与Crohn’s disease在CD4-T中负相关
- HERV→Gene:LTR2B_dup15-chr6与RNASET2表达存在SMR关联
- Gene→Disease:RNASET2表达与Crohn’s disease存在SMR关联
- 共定位:LTR2B_dup15-chr6 eQTL和CD GWAS在rs6939196处共定位,SNP.H4=0.87
- 表观证据:LTR2B_dup15-chr6位于RNASET2上游约1kb增强子样区域,且GeneHancer记录该enhancer和RNASET2互作
- 表达验证:LTR2B_dup15-chr6和RNASET2在CD4-T中正相关,且RNASET2在CD患者CD4-T中下调
rTWAS vs SMR:rTWAS是“表达预测模型”,SMR是“eQTL工具变量检验”
- rTWAS:多SNP预测HERV表达。先训练一个模型
HERV预测表达 = w1×SNP1 + w2×SNP2 + w3×SNP3 + ...,然后拿精神疾病GWAS summary statistics来问这些提高/降低HERV预测表达的SNP,整体上是否也提高/降低疾病风险?- rTWAS的单位是一个表达特征的遗传预测表达,不是单个SNP
- eQTL找哪些SNP影响hERV表达,SMR在其中再找最显著的那个SNP,看同一个SNP是否也影响疾病风险、是否也影响某个gene表达
- 假设某个HERV附近有5个cis-SNP
- rTWAS会训练一个表达预测模型
HERV预测表达 = 0.30×rs1 + 0.12×rs2 - 0.20×rs3 + 0.05×rs4 + 0.18×rs5,然后问这个“由rs1-rs5共同预测出来的HERV表达”是否和疾病风险相关?。不一定要求rs1或rs2单独达到非常强的eQTL显著性。只要这些SNP合起来能预测HERV表达,就可以进入TWAS/rTWAS - SMR通常先找这个HERV的显著eQTL,比如rs3显著调控HERV表达,P<5e-8,然后问
rs3是否也和疾病GWAS风险相关、是否也调控某个gene表达,分别得到variant-eHERV-disease+variant-eHERV-eGene+variant-eGene-disease,三类结果合并后再选
- rTWAS会训练一个表达预测模型
- rTWAS重点探究“某个HERV的遗传预测表达是否和疾病风险相关”,或者说“如果一个人因为遗传因素导致某HERV表达更高/更低,那么他的疾病风险是否也更高/更低”。SMR更直接探究“某个显著调控HERV表达的遗传变异,是否也和gene表达或疾病风险共享遗传信号”
- 局限性:两者都不能直接证明因果
- rTWAS结果不能直接说“HERV表达升高导致疾病”,只能说“由遗传变异预测出的HERV表达变化与疾病风险相关”
- SMR也不能直接说“HERV直接导致疾病”,只能说“调控HERV表达的遗传变异与疾病风险/基因表达存在共享遗传信号”
研究方法和结果总结:
-
单细胞HERV表达定量:UCSC HERV位点级注释+GENCODE gene注释合并,去掉了与基因外显子重叠的HERV位点;cellranger直接计数hERV,只保留unique mapping reads
- 检测到41460个表达HERV位点
-
证明HERV表达相对独立于附近gene:基因组注释、RNA-seq track展示、HERV与最近gene的Spearman相关分析
- 多数表达HERV位于内含子或基因间区,且与附近gene表达相关性低
- 排除“HERV表达只是附近gene转录本的一部分”这个解释
-
识别细胞类型特异HERV:高变HERV筛选、pseudobulk UMAP
- 2045个highly variable HERVs可以使CD4-T、CD8-T、NK、B和myeloid在UMAP中分开
- 得到1936个细胞类型特异HERV
-
把HERV和细胞类型特异表观状态联系起来:Roadmap Epigenomics中15种染色质状态,NCODE H3K27ac/H3K4me3 ChIP-seq定义enhancer/promoter,Fisher富集分析
- 细胞类型特异HERV主要位于active chromatin regions,并富集在对应细胞类型的enhancer/promoter区域
-
构建细胞类型特异HERV cis-eQTL图谱:TensorQTL+pseudobulk分析HERV表达并识别条件独立eQTL
- 共发现3463个条件独立HERV cis-eQTL,涉及1805个HERV和2888个SNP
-
解释HERV eQTL的细胞类型特异性:eSNP与细胞类型特异enhancer的统计富集分析
- 细胞类型特异eSNP富集在对应细胞类型的enhancer中:rs6780016位于CD4-T特异enhancer,调控THE1D_dup769-chr3;rs28894981位于NK相关活性区域,调控LTR9_dup12-chr6
- 作者据此认为HERV cis-eQTL的细胞类型特异性可能由不同细胞类型的表观遗传状态驱动
-
HERV eQTL与免疫疾病GWAS整合:81种免疫相关疾病GWAS,SMR+HEIDI检验
- 发现548个显著HERV-disease pleiotropic associations,涉及196个eHERV和35种疾病
- 自身免疫病最突出,21种自身免疫病中有16种与100个细胞类型特异HERV相关
-
聚焦自身免疫病,尤其是Crohn’s disease:SMR、colocalization、LocusZoom、患者单细胞数据验证
- LTR2B_dup15-chr6在CD4-T中与Crohn’s disease负相关,表达升高对应CD风险降低
- 共定位分析显示LTR2B_dup15-chr6 eQTL和CD GWAS在rs6939196处共享遗传信号
- 患者单细胞数据进一步支持该位点在CD4-T中的疾病相关表达变化
-
HERV-Gene、HERV-Disease、Gene-Disease三类SMR整合:GeneHancer,辅以GeneHancer、ENCODE cCRE、H3K27ac/H3K4me3/DNase-seq轨迹,最后表达相关和患者/健康供体比较
- 识别出168个潜在HERV-gene调控关系,涉及104个HERV和70个gene
- 三类SMR结果重叠后,得到9种自身免疫病、4类细胞、8个HERV和4个gene的候选机制组合
- 代表性机制是CD4-T中的LTR2B_dup15-chr6可能调控RNASET2,并参与Crohn’s disease风险
后续思路
重点——SNP~hERV~疾病:
- 单细胞中大脑皮层的SNP和hERV关系
- 用EPI数据(enhancer-promoter interaction)连接SNP和hERV(SNP在调控元件上,影响不同hERV,联系到疾病中hERV的变化)
- hERV与疾病的因果关系(找mediation):hERV调控转录本导致疾病
- SNP-hERV-疾病:直接overlap/SMR算显著hERV
- 疾病里面失调的TF(SNP影响的TF)同时影响hERV和gene:LTR上结合的TF是哪些
- lncRNA领域:hERV通过miRNA(SNP影响的miRNA)影响基因
- 表观遗传:解释hERV为什么发生变化,作为结果的补充 组蛋白乙酰化/染色质可及性影响比较大,甲基化比较小
这两篇研究存在的不足:它们重点证明“遗传变异能调控hERV,并且这种调控信号与疾病风险相关”,但没有直接回答AD脑组织中哪些hERV在AD和NC之间发生变化(上面的eQTL研究在看hERV实际表达量时是直接用了另外一个数据集做差异表达分析,而不是在主要分析样本中进行),也没有在主要疾病样本中系统分析hERV所在区域的snATAC变化(都是直接用现成的染色质状态图谱)
- 都没说SNP位点是如何影响hERV的
可能的方向
-
表观遗传snATAC-seq:AD中发生变化的hERV/LTR,是否本身就是开放染色质区域、增强子样区域或细胞类型特异调控元件?或者解释hERV为什么变化
上面的eQTL研究已经把细胞类型特异hERV与活跃染色质区域、enhancer/promoter富集联系起来,并提出eSNP富集在对应细胞类型enhancer中,说明细胞类型特异的表观状态可能解释hERV eQTL的细胞特异性。如果同时有AD和NC的数据,就可以直接看AD改变的hERV是否也发生染色质开放性改变
- AD/NC中hERV/LTR区域是否有差异开放峰
- 差异hERV是否落在细胞类型特异开放染色质区域
- hERV/LTR附近是否有H3K27ac、H3K4me1、H3K4me3等enhancer/promoter标记
- hERV/LTR开放性是否和附近gene表达相关(连接hERV/LTR peak和目标gene)
- eQTL对表观遗传:染色质开放区域内是否有GWAS相关SNP位点
-
遗传变异可能通过调控hERV/LTR活性影响gene表达,并与AD风险共享遗传基础:需要每个样本的SNP基因型、hERV和gene表达、AD分组信息
- eQTL分析看SNP和hERV的关系,AD GWAS看AD风险变异是否富集在hERV/LTR区域,enhancer/promoter/EPI注释看AD风险变异是否富集在细胞类型特异开放的hERV/LTR增强子区域
- SMR看HERV-Gene、HERV-Disease、Gene-Disease,hERV和gene是否共享AD风险遗传信号
- mediation:某个因素对疾病的影响,是否有一部分是通过hERV表达变化传递的。比如这两篇文章中
SNP->hERV->疾病的hERV表达就是中间变量,它的研究问题是“这个SNP之所以影响AD,是不是因为它先改变了某个hERV的表达,然后这个hERV再影响AD相关表型?”- SMR说的更多是“SNP-hERV、SNP-AD是否共享同一个遗传信号”,而mediation是
SNP对AD的总影响 = SNP直接影响AD的部分 + SNP通过hERV影响AD的部分 - 相当于构建
AD表型 ~ SNP + hERV表达模型,如果加入hERV表达后,SNP对AD的效应明显变弱,同时hERV本身和AD仍显著相关,就说明hERV可能承接了这个SNP影响AD的一部分效应 - 如果放到
hERV->疾病这个情景下,就是hERV/LTR变化 → gene/transcript表达变化 → AD相关表型/疾病风险,即hERV变化本身未必直接造成病理,而是通过调控附近或远端gene的表达来改变细胞过程
- SMR说的更多是“SNP-hERV、SNP-AD是否共享同一个遗传信号”,而mediation是
-
hERV具体影响的功能验证:
- hERV-gene/gene module共表达
- hERV-TF-gene调控轴:AD风险SNP/AD表观改变→TF结合改变→LTR/hERV活性改变→gene表达改变
- 对AD差异开放的hERV/LTR peak做motif enrichment
- 找出富集TF
- 看这些TF在对应细胞类型中是否AD/NC差异表达
- 看TF target gene是否富集AD相关通路
- 如果AD GWAS SNP落在TF motif上,可以进一步看是否改变TF binding motif
- hERV-miRNA/lncRNA机制:hERV通过miRNA/ceRNA影响gene,需要更多证据,例如hERV转录本结构、miRNA结合位点、miRNA表达、AGO/RISC证据等
- 另一个思路:hERV在脑发育和衰老中整体调控变化,进而在脑部疾病中起作用
- 需考虑多套数据
一些小问题
条件分析的具体原理

- 如果HERV和gene高度相关,比如r=0.9,而且二者边际信号方向相同,那么rZG会从ZH中扣掉很大一部分。此时HERV的条件Z会明显变小,说明它的信号可能主要来自和Gene共享的遗传成分;如果r很低,比如r=0.1,那么扣掉的部分很小。HERV的条件Z接近原来的边际Z,说明HERV更可能是独立信号

- 其中C是预测表达相关矩阵,Z是边际TWAS Z-score向量
- Z向量包含每个feature单独TWAS/rTWAS得到的边际Z值,βjoint是得到每个feature在联合模型中的剩余效应
-
最后的joint P由前面几个变量推到而来

为什么“条件分析后的局部区域图”有时灰点高有时蓝点高

- 所以这种情况可能提示:该区域不是一个简单信号,而可能有多个遗传效应叠在一起,有的使疾病风险降低,有的使升高
- 也要注意纵轴是信号强度而不是方向,有时条件化后SNP效应方向可能已经改变,但图上只显示P值变小或变大,不说明它的方向仍然和原来一致
- 不要只看某一个SNP的蓝点高低,而要看整个区域的模式:
- 如果原始峰附近大多数蓝点都低于灰点,说明被条件化的gene/HERV预测表达解释了相当一部分GWAS信号
- 如果蓝点和灰点差不多,说明这些表达特征对原始GWAS峰解释有限
- 如果蓝点在某些位置高于灰点,尤其是形成一个新的峰,说明可能存在另一个独立遗传信号,或者原始信号中有方向相反的成分被条件化后显现出来

fine-mapping(FOCUS)的具体原理

后验纳入概率(posterior inclusion probability, PIP):PIPj=P(featurej是真正解释该locus信号的feature∣观察到的TWAS Z值和相关性)
- PIP不是P值。P值回答“这个feature是否和疾病相关”;PIP回答“在这个区域所有候选feature中,它是不是解释信号的候选原因之一”


可信候选集合(credible set):把PIP从高到低排序,累加到某个阈值,比如90%,得到一组最值得后续验证的feature

-
例子1:HERV信号强,且和其它feature相关性低
HERV1 Z=7.0 GeneA Z=2.0 GeneB Z=1.5 cor(HERV1,GeneA)=0.10 cor(HERV1,GeneB)=0.05这时HERV1的信号强,而且不能被附近gene的预测表达相关性解释。FOCUS可能给:
HERV1 PIP=0.95 GeneA PIP=0.02 GeneB PIP=0.01解释:HERV1很可能是这个区域的主解释feature
-
例子2:HERV和GeneA都强,但二者预测表达高度相关
HERV1 Z=7.0 GeneA Z=6.8 GeneB Z=1.2 cor(HERV1,GeneA)=0.97这时FOCUS很难区分HERV1和GeneA,因为它们的预测表达几乎一样。它可能给:
HERV1 PIP=0.50 GeneA PIP=0.47 GeneB PIP=0.01解释:这个区域确实有表达介导信号,但目前分辨率不足,不能确定是HERV1还是GeneA
- 相关性越复杂,FOCUS越可能把概率分散到多个feature,而不是集中给某一个HERV
-
例子3:HERV显著,但它的信号形状更像被GeneA牵连出来
HERV1 Z=5.5 GeneA Z=7.5 GeneB Z=2.0 cor(HERV1,GeneA)=0.85如果GeneA是真正解释feature,那么HERV1因为和GeneA相关,也会出现不弱的TWASZ。FOCUS可能判断“GeneA作为主feature更能解释整体Z模式”,于是给:
GeneA PIP=0.80 HERV1 PIP=0.15 GeneB PIP=0.03解释:HERV1虽然rTWAS显著,但它更像是被GeneA牵连出来的信号
SMR的具体原理
原始SMR方法是把GWAS summary data和eQTL summary data整合,用来找“基因表达水平和复杂性状是否因为共享遗传变异而相关”的方法,换句话说,就是检验SNP对表型的效应是否可能由表达介导
- 在这里,gene expression被替换或扩展成HERV expression:如果同一个SNP既强烈影响HERV表达,又影响疾病风险,那么SMR就会认为这个HERV和疾病之间存在遗传层面的关联



- 如果P_SMR显著,就说明这个HERV和疾病之间存在遗传层面的关联
- 调控HERV_X表达的遗传信号,同时也和Crohn’s disease风险相关,但不能说“HERV_X表达升高导致Crohn’s disease”
SMR显著有两种常见解释:
- 共享因果变异/pleiotropy:同一个SNP rsA既影响HERV表达,又影响疾病风险,此时SMR想要的信号比较可信
- linkage/LD牵连:rsA真正影响HERV表达,rsB真正影响疾病风险,rsA和rsB因为LD很强,所以看起来像同一个信号,这时SMR也可能显著,但解释就变弱了,因为HERV表达和疾病风险可能不是同一个因果SNP驱动,而只是两个相邻信号被LD绑在了一起
这时就需要HEIDI:

染色质状态分布如何得到
在2024 NC–schERV+eQTL中,作者直接下载了Roadmap Epigenomics Project中5类PBMC相关细胞的15-state chromatin states,然后把HERV位点或eSNP位置和这些染色质状态区间做重叠统计
染色质状态:把整个人类基因组按表观修饰组合切成很多小区间,并给每个区间贴一个功能标签(来自多个组蛋白修饰信号组合)
- 例如某个基因组区间
chr1:100000-100200,如果它在CD4-T细胞中有强H3K4me3和H3K27ac信号,且靠近TSS,就可能被标成Active TSS - 另一个区间
chr1:500000-500200,如果它有H3K4me1和H3K27ac,但没有明显H3K4me3,就可能被标成Enhancer - 再比如有H3K27me3,就可能被标成Polycomb相关抑制状态;如果几乎没有活性标记,就可能是Quiescent/Low
Roadmap Epigenomics使用的是ChromHMM。它的核心输入是不同细胞/组织中的组蛋白修饰ChIP-seq信号
| 组蛋白标记 | 常见含义 |
|---|---|
| H3K4me3 | 活跃启动子、TSS附近 |
| H3K4me1 | enhancer,尤其是候选增强子 |
| H3K36me3 | 转录延伸区域,常见于gene body |
| H3K27me3 | Polycomb介导的抑制区域 |
| H3K9me3 | 异染色质、重复序列相关抑制区域 |
- Polycomb抑制区域,一类进化上高度保守的转录抑制因子
之后ChromHMM会把基因组切成固定大小的小窗口,常见是200bp左右,然后判断每个窗口中这些标记是否存在,根据每个窗口中这些标记的组合模式,把全基因组窗口归成15类状态。例如:
- H3K4me3强 + 靠近已知TSS → Active TSS
- H3K4me1强 + 远离TSS → Enhancer
- H3K36me3强 + 位于基因体 → Transcription/Strong transcription
- H3K27me3强 → Repressed PolyComb
- H3K9me3强 → Heterochromatin/ZNF genes & repeats
在这篇文章中,作者先识别细胞类型特异HERV,然后对每一类HERV看“这些HERV在对应细胞类型的基因组里,更常落在哪些染色质状态中?”。例如一个CD8-T特异表达的HERV,如果它的基因组坐标和CD8-T细胞Roadmap注释里的Enhancer区间重叠,那么这个HERV在CD8-T中就被认为落在enhancer状态
15种状态包括:
Active TSS
Flanking Active TSS
Transcription at gene 5' and 3'
Strong transcription
Weak transcription
Genic enhancers
Enhancers
ZNF genes & repeats
Heterochromatin
Bivalent/Poised TSS
Flanking Bivalent TSS/Enh
Bivalent Enhancer
Repressed PolyComb
Weak Repressed PolyComb
Quiescent/Low
通常作者说active chromatin regions时,主要指前面那些有启动子、转录或增强子特征的状态,例如Active TSS、Flanking Active TSS、Strong/Weak transcription、Genic enhancers、Enhancers等。相对地,Heterochromatin、Repressed PolyComb、Quiescent/Low更偏沉默或低活性状态
总结:表观遗传标记
| 标记 | 常见提示的染色质/调控含义 | 可以怎么理解 |
|---|---|---|
| H3K4me3(常用) | 活跃启动子、TSS附近 | 如果一个区域H3K4me3很强,通常说明这里像转录起始区域,常见于gene promoter |
| H3K4me1(常用) | enhancer候选区域 | 单独H3K4me1常提示增强子潜力;如果再叠加H3K27ac,更像活跃enhancer |
| H3K27ac(常用) | 活跃enhancer和活跃promoter | 区分“准备好但不一定活跃”的enhancer和“正在活跃”的enhancer很常用;文章中看HERV是否落在活性enhancer时经常会参考它 |
| H3K9ac | 活跃启动子/开放染色质 | 与转录活跃区域相关,常和H3K4me3一起出现在活跃promoter附近 |
| H3K14ac | 活跃转录区域 | 也是乙酰化活性标记,常提示染色质较开放、转录因子更容易进入 |
| H3K4me2 | promoter和部分enhancer | 介于H3K4me1和H3K4me3之间,常见于启动子附近,也可出现在调控元件 |
| H3K36me3 | 转录延伸区域/gene body | 常出现在正在被转录的基因主体区域,表示RNApolII已经通过并进行延伸 |
| H3K79me2 | 活跃转录相关gene body | 常与转录活跃基因内部区域相关,可辅助判断转录延伸 |
| H4K20me1 | 转录区域、细胞周期相关调控 | 在一些ChromHMM扩展模型中用于辅助识别转录区域或特定染色质状态 |
| H3K27me3(常用) | Polycomb介导的抑制区域 | 常见于发育调控基因或被Polycomb压制的区域;不是永久沉默,更像“被压住” |
| H3K9me3(常用) | 异染色质、重复序列、强抑制区域 | 常见于重复序列、卫星DNA、转座元件富集区域;ENCODE也特别指出H3K9me3富集于重复区域,分析时多重比对会更复杂 |
| H3K9me2 | 抑制性异染色质/大范围低活性区域 | 和H3K9me3类似但常更广泛,提示较压缩、低转录活性的染色质 |
| DNA甲基化/5mC(常用) | 启动子甲基化常与转录抑制相关;gene body甲基化含义更复杂 | 如果HERV/LTR启动子甲基化降低,可能提示转座元件去抑制;但甲基化位置不同,解释不同 |
| 5hmC | DNA去甲基化中间状态或特定组织活性标记 | 在神经系统中特别常见,不能简单等同于5mC抑制 |
| ATAC-seq信号(常用) | 开放染色质 | 说明该区域染色质更容易被Tn5进入,常提示可能有promoter/enhancer/TF结合活性 |
| DNase-seq信号(常用) | 开放染色质 | 和ATAC-seq类似,表示该区域更容易被DNaseI切割,常用于识别调控元件 |
| CTCF | 绝缘子、TAD边界、染色质环锚点 | 如果HERV附近有CTCF,可能提示它处在染色质结构边界或loop相关区域 |
| Cohesin/RAD21/SMC3 | 染色质环、增强子-启动子接触 | 常和CTCF一起解释3D染色质结构 |
| RNApolII/Pol2 | 转录机器占据 | Pol2在启动子附近富集提示转录起始或暂停;在gene body中延伸提示正在转录 |
| TFChIP-seq | 特定转录因子结合 | 比如NF-κB、STAT、IRF等结合可提示免疫/干扰素调控;对HERV免疫相关机制解释很有用 |
按基因组类型分组:
| 类型 | 常见标记 | 主要含义 |
|---|---|---|
| 活跃启动子 | H3K4me3、H3K9ac、H3K27ac、Pol2 | 这里可能是转录起始点 |
| 活跃增强子 | H3K4me1+H3K27ac、ATAC/DNase开放 | 这里可能增强附近基因或HERV转录 |
| 潜在/poised增强子 | H3K4me1高、H3K27ac低 | 有增强子潜力,但当前细胞类型中不一定活跃 |
| 转录延伸 | H3K36me3、H3K79me2、Pol2gene body | 这里可能是正在被转录的基因主体 |
| Polycomb抑制 | H3K27me3 | 发育相关或可逆抑制区域 |
| 异染色质/重复序列抑制 | H3K9me3、H3K9me2、DNA甲基化 | 常见于转座元件、重复序列和强抑制区域 |
| 染色质结构 | CTCF、RAD21、SMC3 | 解释边界、loop和增强子-启动子空间接触 |
具体来说
| 问题 | 最推荐看的标记 |
|---|---|
| 这个HERV是否像enhancer? | H3K4me1+H3K27ac+ATAC/DNase |
| 这个HERV是否像promoter? | H3K4me3+H3K27ac+RNApolII |
| 这个HERV是否被转座元件沉默机制压制? | H3K9me3+DNA甲基化 |
| 这个HERV是否只是落在宿主gene转录区域? | H3K36me3+RNA-seq方向+gene annotation |
| 疾病中是否发生HERV去抑制? | H3K9me3下降、DNA甲基化下降、ATAC/H3K27ac升高、HERV表达升高 |
polyA尾相关
简单回顾RNA的成熟过程:
-
DNA上有一个promoter,转录因子、Mediator、RNA Pol II等复合体被招募到这里,然后RNA Pol II开始沿着DNA模板合成RNA,这时产生的是前体RNA
DNA转录方向为RNA链沿5’→3’方向延伸,对应DNA模板链的3’→5’方向
- RNA刚开始出来时,5’端会加cap;如果有内含子,会发生剪接,把intron去掉,把exon连起来
- 当RNA Pol II转录到一个可用的polyadenylation signal,比如AAUAAA附近时,CPSF、CstF等复合体会识别这个区域,然后在下游某个位点把RNA切开,poly(A)polymerase再在新的3’端加上一串A,也就是polyA尾
- 总的来说,真核mRNA的3’端形成通常包括“先内切剪切,再polyadenylation加尾”两个步骤,所以polyA尾不是“转录时自动加上”的,而是需要RNA上有合适的3’端加工信号,并且相关加工复合体真的在这里工作
promoter和enhancer的本质差别是什么:
- promoter的主要任务是生产一个相对完整的RNA转录本。它通常位于基因转录起始位点附近,方向比较明确,和RNA Pol II起始、5’cap、剪接、3’端polyA加工等流程配合得比较好
- enhancer的主要任务不是生产RNA,而是增强别的promoter转录。它通常是转录因子结合密集的调控区域,可以通过染色质环化接触目标promoter,帮助目标基因表达增强。因为enhancer附近有转录因子、p300/CBP、Mediator、RNA Pol II等活跃转录相关因子,所以enhancer本身也可能被RNA Pol II“顺手转录”,产生eRNA
enhancer表达出来的RNA不带polyA尾?:大多数典型eRNA不稳定、常不带polyA尾;但如果enhancer区域产生的是更像lncRNA的长转录本,它也可以带polyA尾
- promoter启动的正向转录更容易延伸成完整RNA,而很多enhancer或反向转录更容易早停、降解
- enhancer转录常常是“局部短距离转录”:RNA Pol II在enhancer附近起始后,常常只转录一小段,很快终止,这类RNA往往短、低丰度、不稳定,容易被核内RNA降解系统清除。很多典型eRNA表现为bidirectional transcription,也就是从enhancer中心向两个方向都转录
- 它们不一定有合适的3’端加工结构:很多enhancer产生的eRNA没有形成这种高效3’端加工结构,所以不会积累成稳定polyA RNA
- promoter附近有保护长转录本的机制,enhancer不一定有:U1snRNP除了参与剪接,也能抑制过早cleavage/polyadenylation,帮助RNA Pol II继续转录更长的基因
- 一个DNA区域可以有enhancer活性,同时也可能具备类似promoter的能力。如果这个区域产生的RNA满足“转录方向比较固定、RNA长度较长、有剪接结构、下游有可用polyA signal、RNA能逃过快速降解、3’端加工复合体能完成剪切和加尾”,它就可能带polyA尾。这种RNA就不像典型短eRNA,而更像lncRNA
人体ncRNA什么情况下会发生ployA加尾:关键不是“它是不是ncRNA”,而是“它是不是走RNA Pol II长转录本加工路线”
- 容易带polyA尾的ncRNA:很多lncRNA、antisense RNA、pseudogene transcript、部分TE/HERV来源转录本,都是RNA Pol II转录的。如果它们有合适的polyA signal,就可以像mRNA一样发生3’端剪切和polyA加尾,所以这些RNA虽然“不编码蛋白”,但它们可以是polyadenylated ncRNA
- 很多小RNA不是典型RNA Pol II长转录本,比如rRNA、tRNA、snRNA、snoRNA、miRNA、piRNA等。它们有各自的加工和成熟机制,通常不靠经典mRNA式polyA尾来稳定
- 某些非polyA RNA在降解过程中可能会被加上很短的A尾,这种短A尾有时是降解标记,不是成熟稳定RNA的polyA尾
在hERV中:
- 如果一个HERV/LTR区域有H3K27ac、H3K4me1、转录因子结合,并增强附近基因表达,那么它可以被看作enhancer样调控元件,这时即使它产生一些短eRNA,也可能不带polyA尾
- HERV/LTR像alternative promoter:如果LTR自己作为启动子,向下游gene exon转录,并且和宿主基因发生剪接,那么它就可能生成一个HERV-gene嵌合转录本,这种转录本后面往往可以使用宿主基因原本的polyA位点,所以它更可能带polyA尾
- HERV只是位于某个宿主转录本内部
//todo 另外的polyA加尾机制(和LTR相关) //polyA尾降解机制(和炎症相关)
0606可变剪接网课
可变剪接在细胞分化、组织分化、发育中广泛存在。在疾病情况下,剪接改变,产生不正常转录本推动疾病发生发展,可作为疾病标志物或治疗靶点。在癌症细胞中有很多癌症特异转录本,作为肿瘤特异性抗原,可作为CART等方法精准治疗的靶点
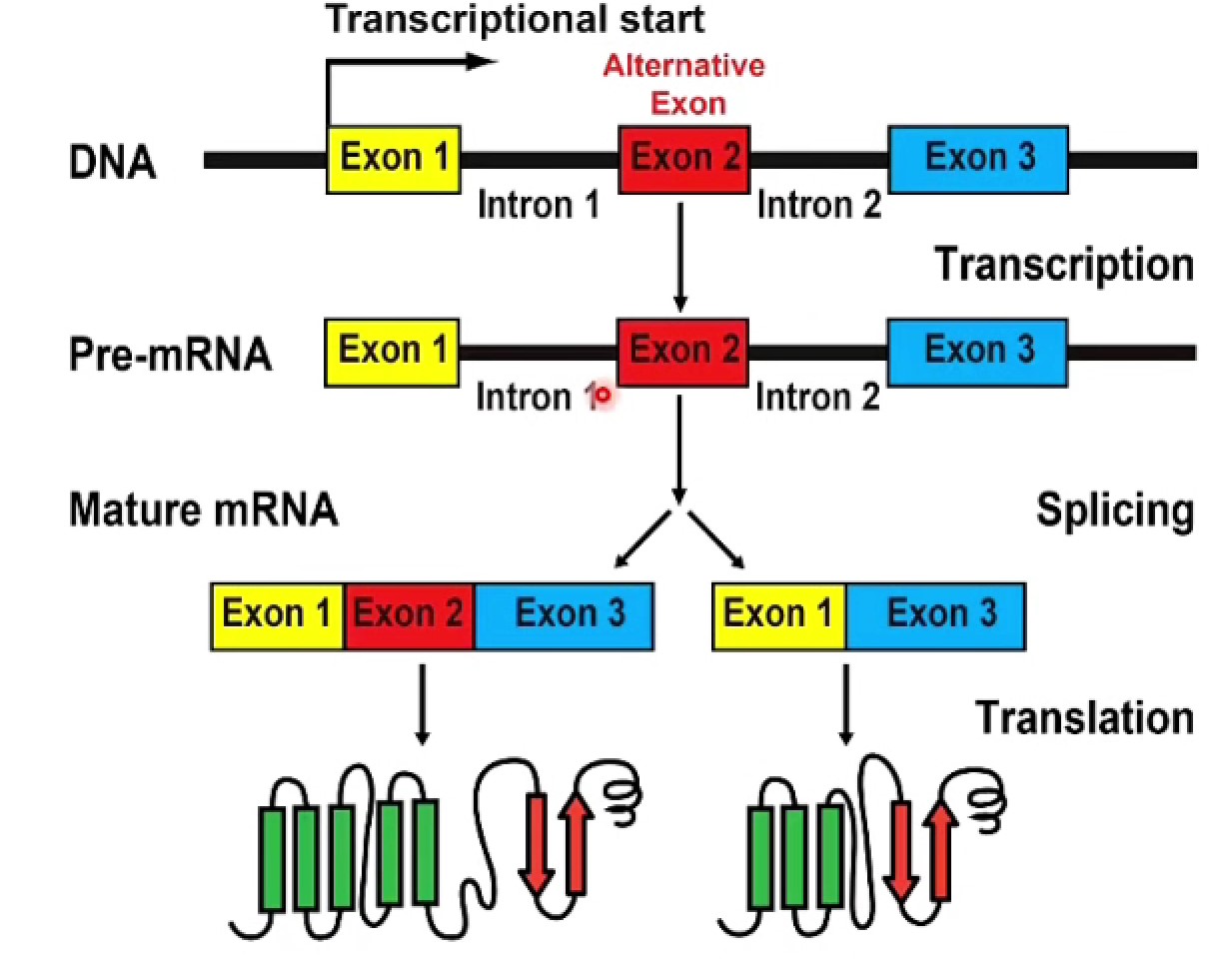
- 最简单的例子:exon2可以被/不被剪接进成熟mRNA中,产生多种剪接异构体(isoform)
- isoform可以有很多种,比如KCNMA1就有>40种
- 几乎所有的基因都能产生isoform
一个重要的问题:发现/定量转录本——转录组学(transcriptomics)
- 最早:1991年在人类基因组计划中创新的技术——expressed sequence tags(ESTs),通过分析每个基因产生的mRNA(cDNA)来测量基因(转录本)结构,少部分是全长,大部分都是片段(fragments)
- 20世纪初:通过大量比对EST和基因组,发现转录本水平上的变异
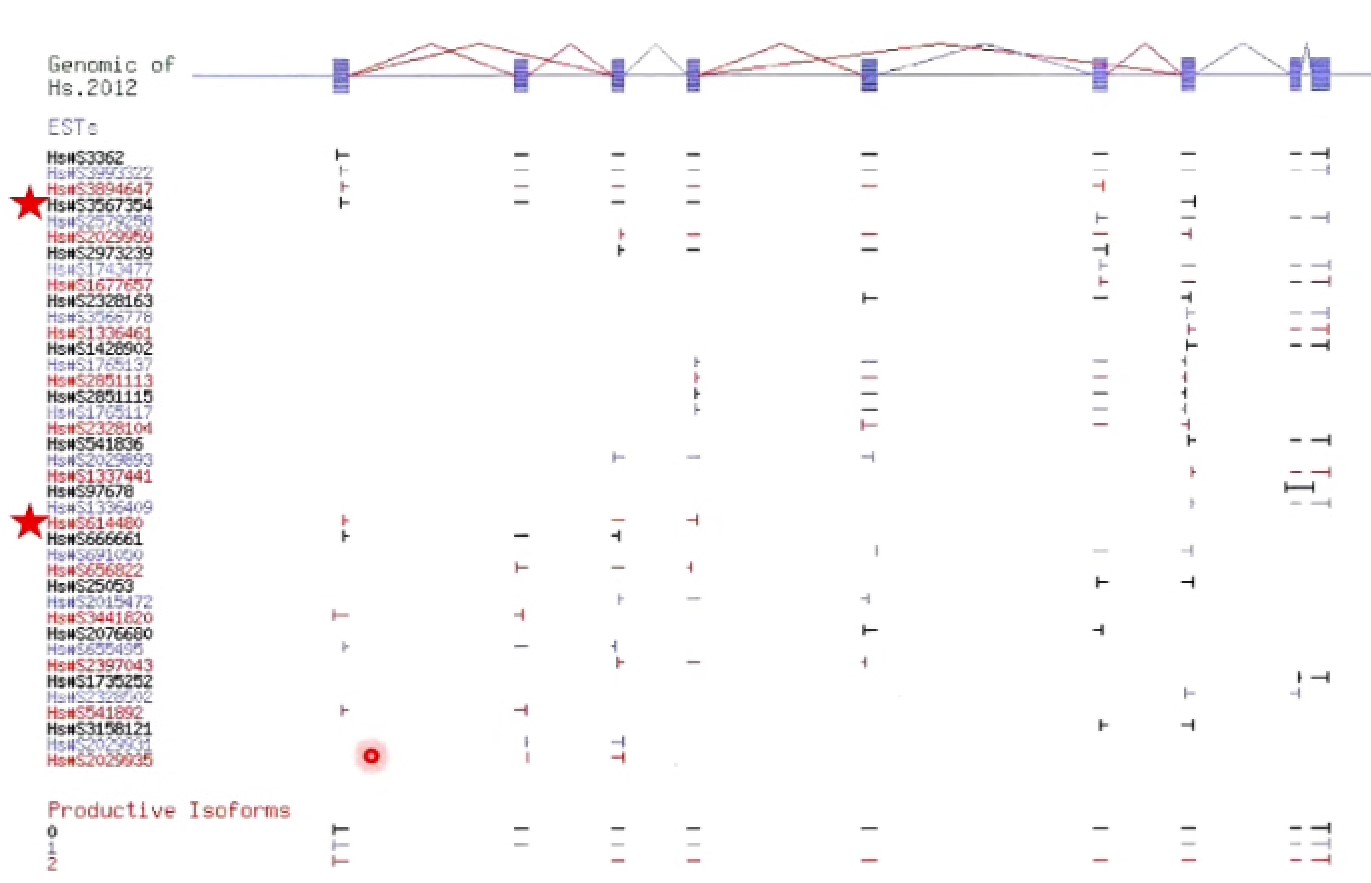
EST组装的问题:
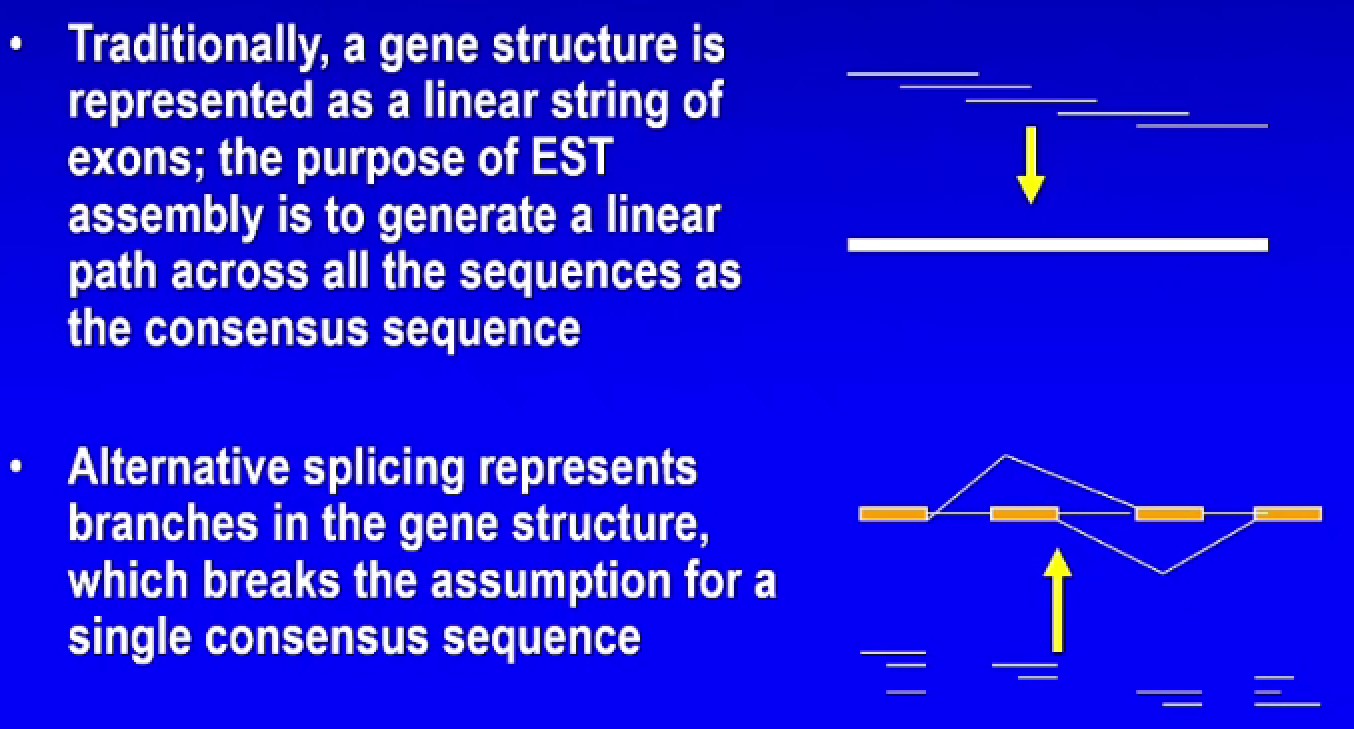
- 如果有一组序列是来自同一个基因,怎么产生一个线性的途径包含所有的序列片段(consensus sequence)
- 考虑到可变剪接里面基因结构会出现分支,consensus sequence的方法就不行了
-
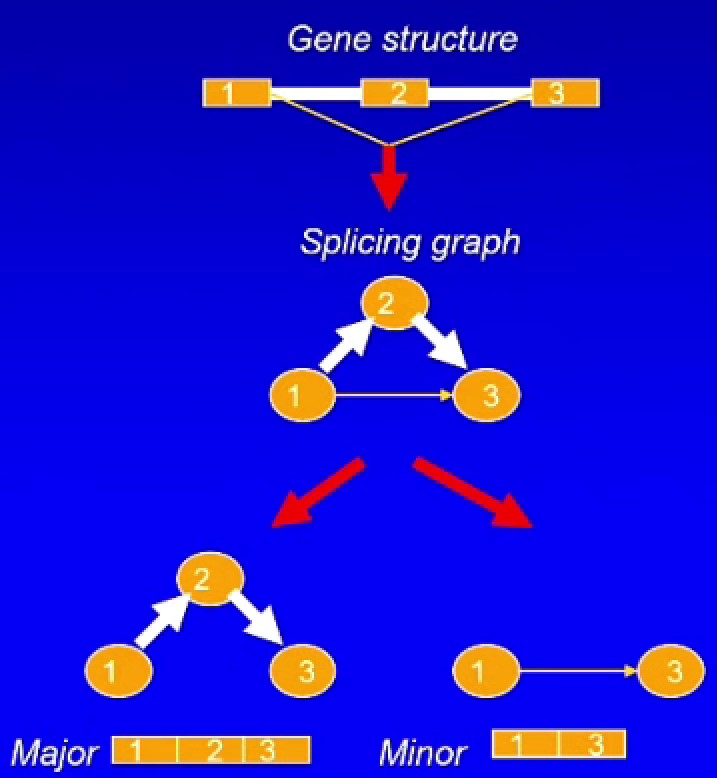
splice graph:每个外显子都是一个节点,剪接事件是边,在边上放不同权重(如果看到的次数多权重就大),isoform就是遍历这个有向无环图得到的片段组合
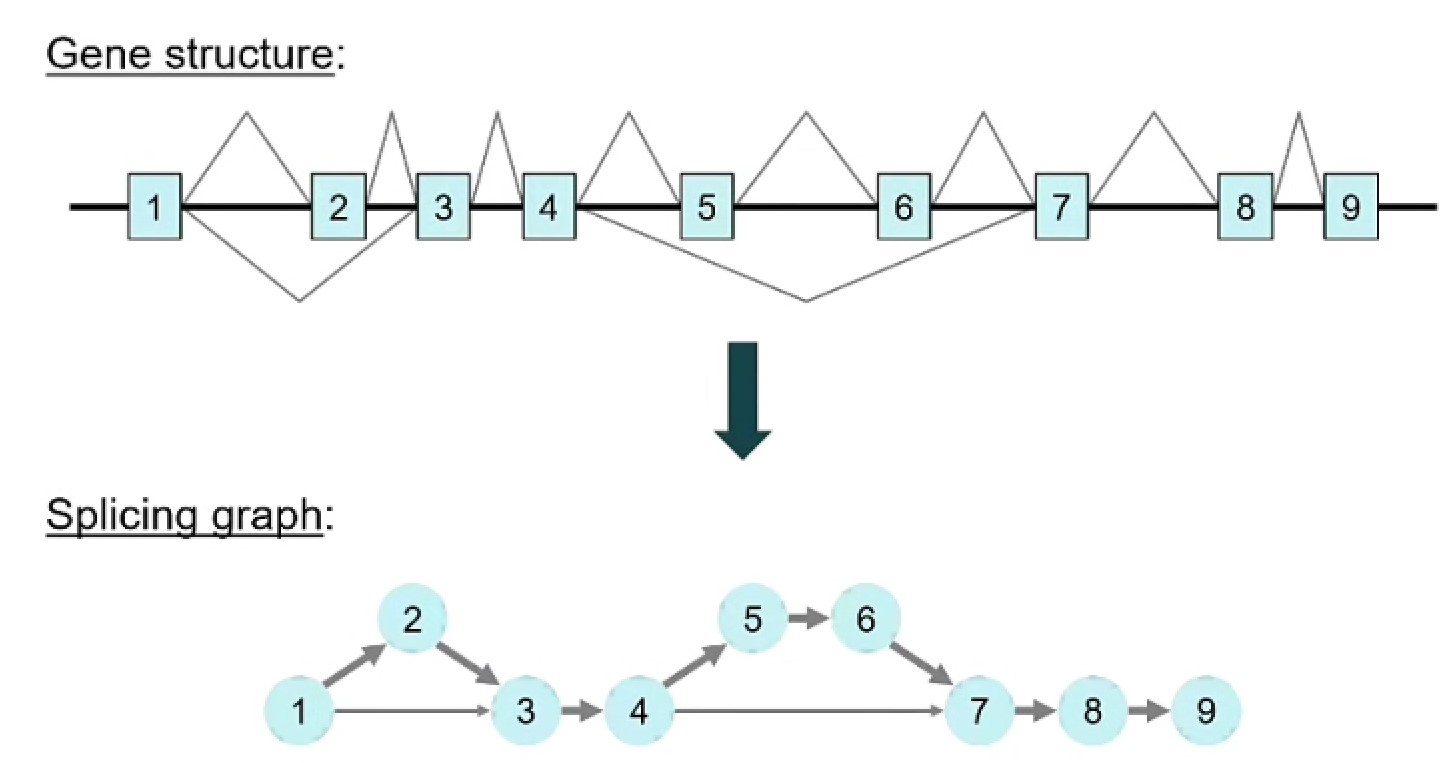
通过动态规划算法来组装splice graph:
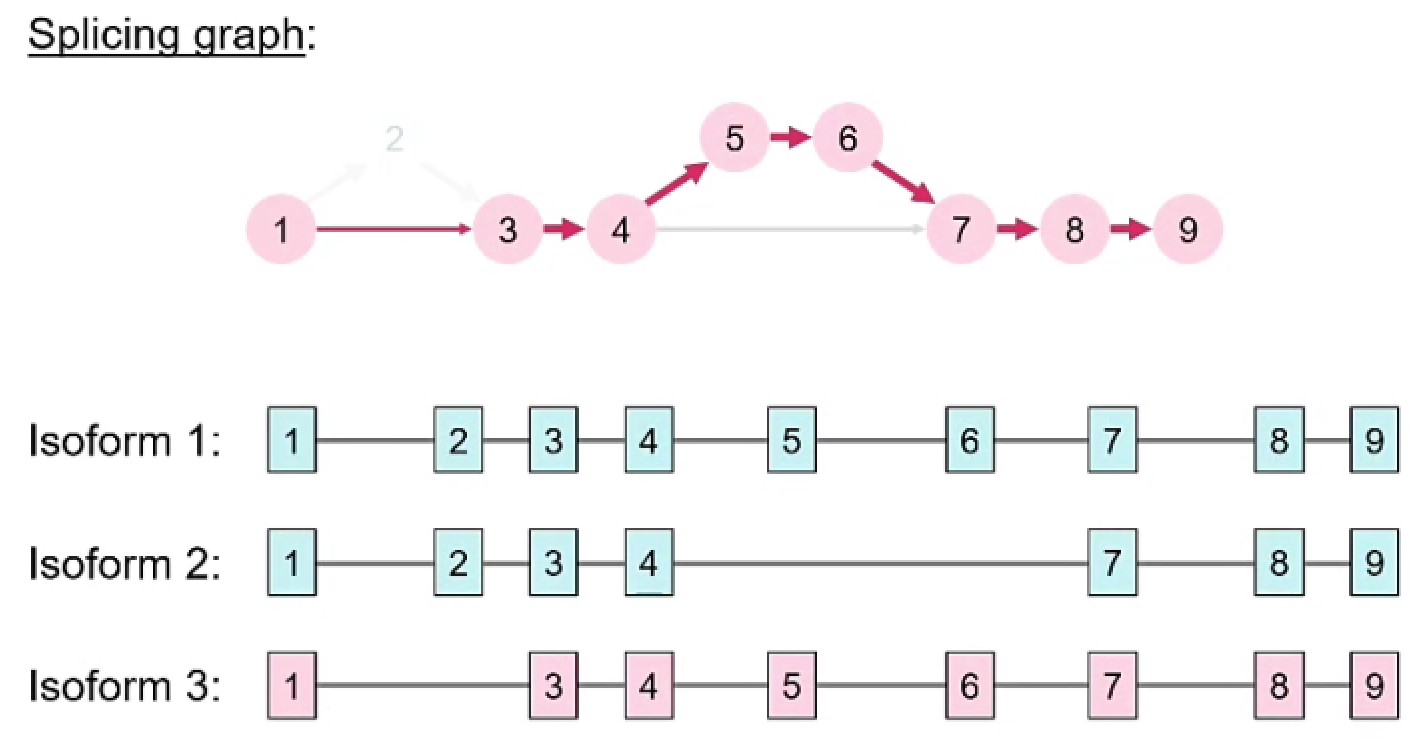
EST sequence通量有限,15年才得到6-700w条。后面出现了微阵列基因芯片、短读长RNA-seq、长读长RNA-seq,当测序深度足够深时就能看到多种不同的isoform——可能有一些外显子在某个细胞系中特异表达。rMATS等工具快速分析大量可变剪接方式,进而用深度学习进行预测
- 短读长很难找全长转录本
- 长读长产生>10kb的reads,直接可以读取完整转录本
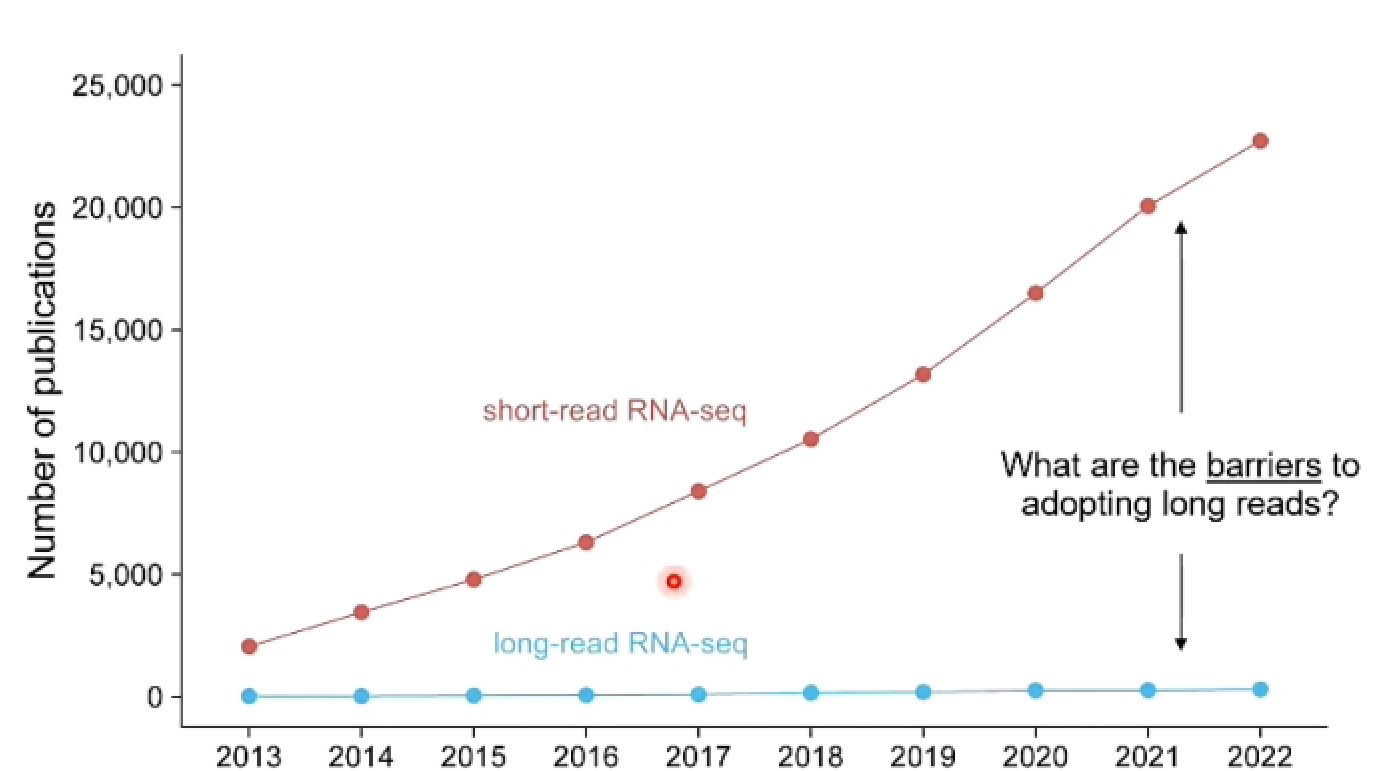
- 为什么大家都不用长读长?
- 长读长序列准确度要低:短读长99.9% 长读长88-89%(现在97.5%左右)
- 长读长成本高:1000$ 短读长2×108 长读长4×105-5×106
- ESPRESSO:通过一些算法来对错误率较高的转录本进行分析和定量
- TEQUILA-seq:提高对基因的coverage
应用——罕见病诊断:全基因组测序发现致病基因突变,大多数都是单基因突变,但基因组里突变很多,如何定位致病突变很难——只有30%患者能得到遗传学诊断
RNA-seq可以更好确定致病突变:不仅知道基因组上哪里突变,还知道这个突变对转录本的影响
在遗传分析上,短读长RNA-seq的问题:不知道突变(突变的外显子)是从母本还是父本上来的。而长读长可以得到单倍体分辨率的转录本,确定突变来源
- 问题:长读长测序太贵了——一个样本2000-3000$
- 解决方法:不测全转录组——如果已知疾病,就只测疾病相关基因——靶向测序,TEQUILA技术获取靶向目标基因的引物,这样测得的reads就大量富集在目标基因上,成本100$/样本
剪接位点突变可以导致内含子剪接改变:
- 内含子保留(intron retention):一种可变剪接形式,指在mRNA剪接过程中,内含子未被剪除而保留在成熟mRNA中的现象
- 内含子多聚腺苷酸化(Intronic polyadenylation,IPA):一种可变剪接形式,基因使用内含子poly (A)位点从而产生截短转录本的现象
- 生物学解释:U1 snRNP识别剪接位点,突变后,U1 snRNP无法识别剪接位点导致内含子中的polyA暴露,让转录本截短
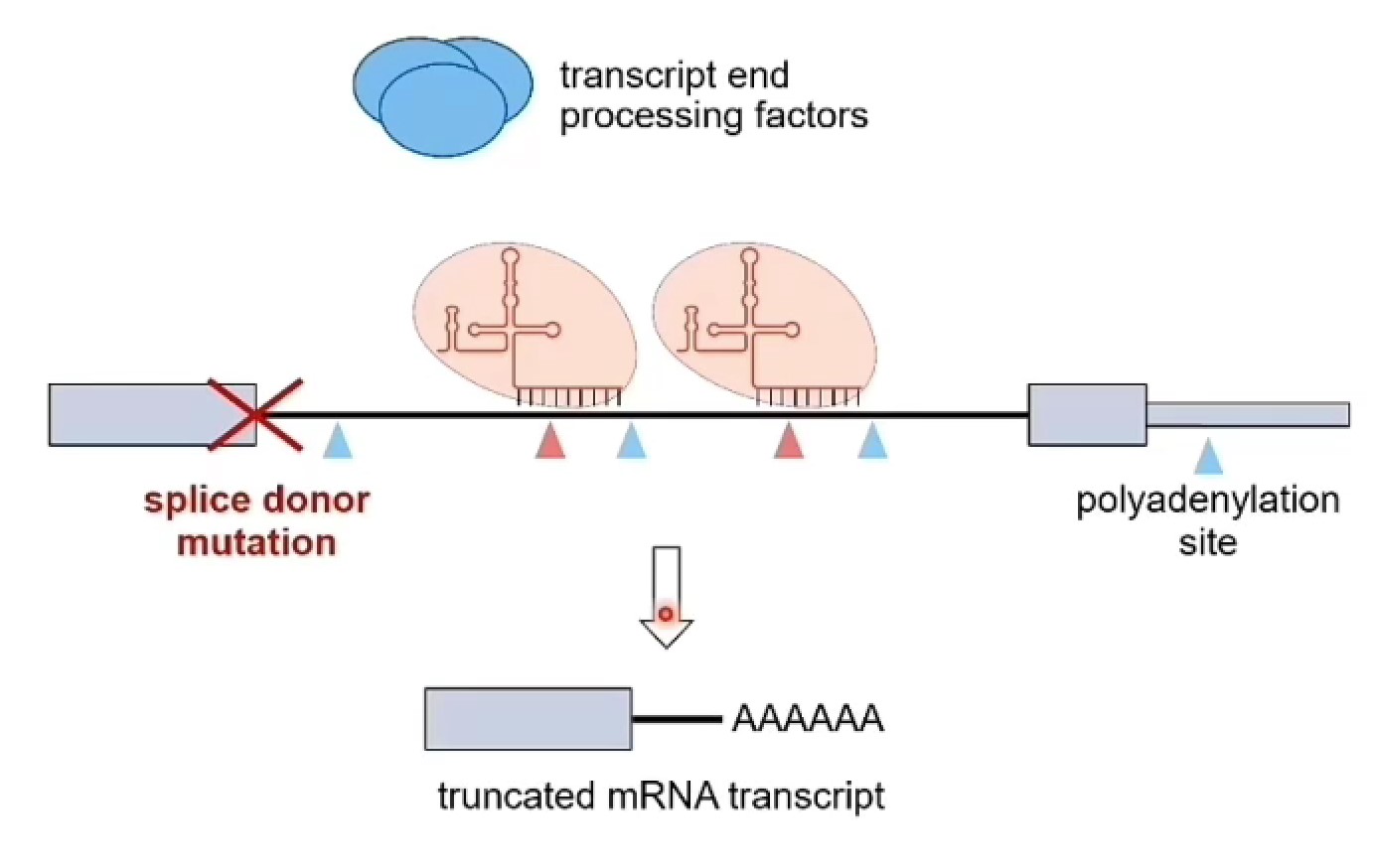
剪接供体位点突变(splice donor mutation)导致很多遗传疾病:是剪接突变的一种,指的是发生在内含子5’端剪接位点的突变,也就是外显子和下游内含子交界处附近的剪接信号被破坏
外显子 | 内含子
...AG | GT...(DNA层面)
...AG | GU...(转录成RNA后)
- 其中内含子开头的GT/RNA中的GU就是典型的splice donor site,也叫5’ splice site
- 常见写法比如
c.123+1G>A/c.123+2T>C,这里+1、+2通常就是外显子后面内含子起始的第1、第2个碱基 - 相对地,splice acceptor mutation是发生在内含子3’端、靠近下一个外显子的剪接受体位点,经典信号通常是内含子末端的
AG,写法常见如c.124-1G>A/c.124-2A>G
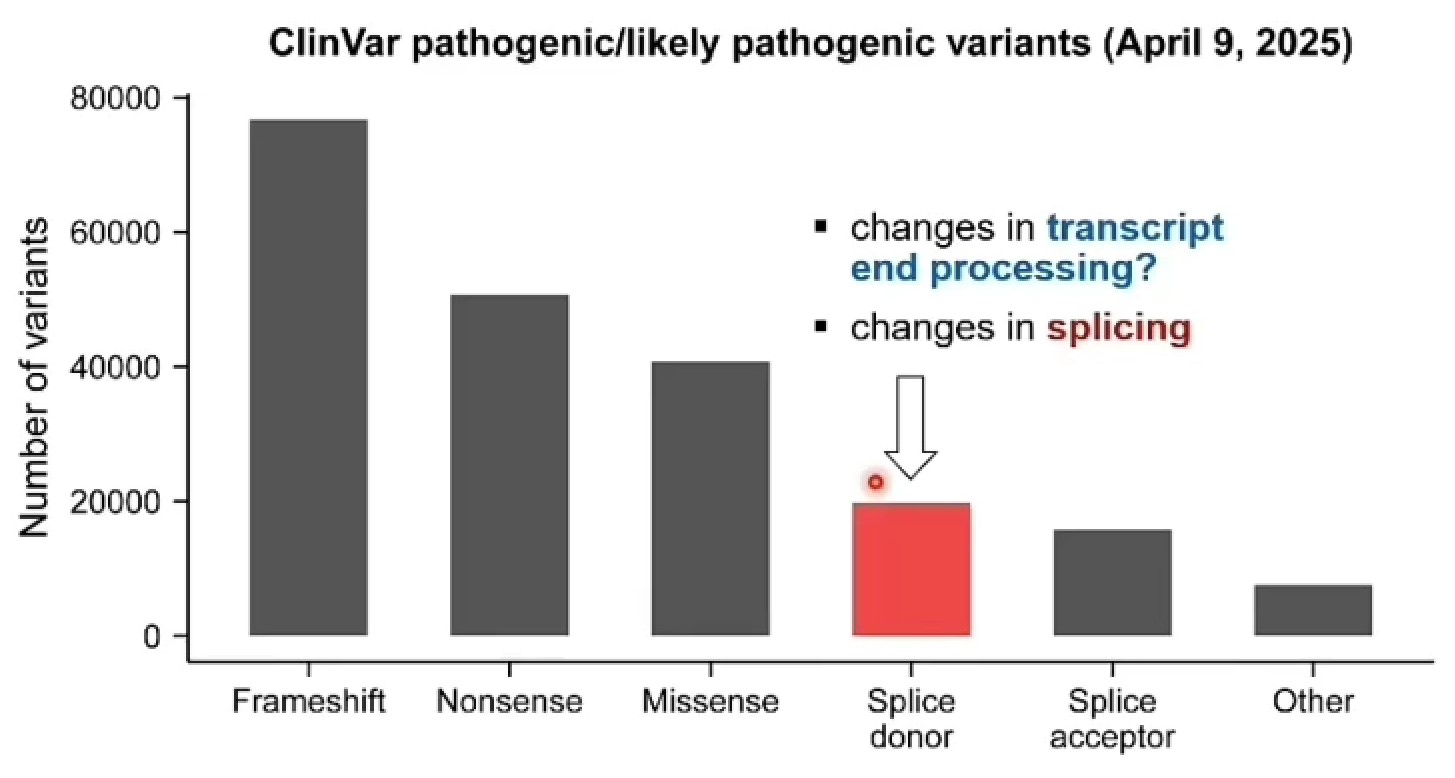
总结:
- 个人基因组的临床应用受到功能性变异解释的限制
- 长读长RNA-seq可以捕捉遗传变异对分子表型的顺式效应
- 成本一直是主要瓶颈
- 开发一种靶向策略,实现对疾病相关基因的深度、全长测序(每位患者样本约100美元)
- 已知致病变异可能产生意料之外的后果(例如,剪接供体突变→内含子内多聚腺苷酸化)
- 影响基因调控的致病变异可能被漏检,包括深内含子变异、同义变异和结构变异
- 需要长读长技术来捕捉我们基因组中完整的变异谱(例如重复区域)
一些问题:
- 区分真正有功能的可变剪接和剪接噪音最关键的标准:细胞组织特异、跨物种保守,具体功能还需实验验证。这个问题现在也没有很明确的解决方法,可以不知道每个isoform的生物学功能 也可以上作为疾病标志物
- 长读长数据和其它数据类型联合运用:scRNA、短读长数据(因为长读长错误率高,用短读长验证,但同一样本测两次成本很高,现在因为长读长准确率提高所以一般就只用长读长了)
- 肿瘤新抗原筛选:DNAseq+RNAseq互补,DNA看到体细胞突变,但数量有限,RNA看转录本-蛋白-抗原层面
hERV位点和基因的重叠
hERV位点和基因的重叠,是不是重叠多的表达高
# hERV-gene重叠注释表
herv_df <- fread("/public/home/GENE_proc/wth/GSE174367/0530test/data/hERV_locus.bed", header = FALSE)
colnames(herv_df) <- c("chr", "start0", "end", "herv_id", "score", "strand")
herv <- GRanges(
seqnames = herv_df$chr,
ranges = IRanges(start = herv_df$start0 + 1, end = herv_df$end),
strand = herv_df$strand
)
mcols(herv)$herv_id <- herv_df$herv_id
mcols(herv)$herv_len <- width(herv)
gtf <- import("/public/home/wangtianhao/Desktop/STAR_ref/gencode.v49.annotation.gtf")
genes <- gtf[gtf$type == "gene"]
exons <- gtf[gtf$type == "exon"]
std_chr <- paste0("chr", c(1:22, "X", "Y"))
herv <- keepSeqlevels(herv, intersect(seqlevels(herv), std_chr), pruning.mode = "coarse")
genes <- keepSeqlevels(genes, intersect(seqlevels(genes), std_chr), pruning.mode = "coarse")
exons <- keepSeqlevels(exons, intersect(seqlevels(exons), std_chr), pruning.mode = "coarse")
herv_base <- data.frame(
herv_id = mcols(herv)$herv_id,
chr = as.character(seqnames(herv)),
start = start(herv),
end = end(herv),
strand = as.character(strand(herv)),
herv_len = width(herv)
)
gene_hits <- findOverlaps(herv, genes, ignore.strand = TRUE)
gene_ol <- data.frame(
herv_i = queryHits(gene_hits),
gene_i = subjectHits(gene_hits)
)
gene_ol$herv_id <- mcols(herv)$herv_id[gene_ol$herv_i]
gene_ol$gene_id <- genes$gene_id[gene_ol$gene_i]
gene_ol$gene_name <- genes$gene_name[gene_ol$gene_i]
gene_ol$gene_type <- genes$gene_type[gene_ol$gene_i]
gene_ol$gene_strand <- as.character(strand(genes))[gene_ol$gene_i]
gene_ol$herv_strand <- as.character(strand(herv))[gene_ol$herv_i]
gene_ol$same_strand <- gene_ol$gene_strand == gene_ol$herv_strand
gene_ol$overlap_bp <- width(pintersect(herv[gene_ol$herv_i], genes[gene_ol$gene_i], ignore.strand = TRUE))
gene_sum <- gene_ol %>%
group_by(herv_id) %>%
summarise(
n_gene_overlap = n_distinct(gene_id),
n_protein_coding_gene = n_distinct(gene_id[gene_type == "protein_coding"]),
same_strand_gene_n = n_distinct(gene_id[same_strand]),
opposite_strand_gene_n = n_distinct(gene_id[!same_strand]),
gene_overlap_bp_max = max(overlap_bp),
gene_list = paste(unique(gene_name), collapse = ";"),
.groups = "drop"
)
exon_hits <- findOverlaps(herv, exons, ignore.strand = TRUE)
exon_ol <- data.frame(
herv_i = queryHits(exon_hits),
exon_i = subjectHits(exon_hits)
)
exon_ol$herv_id <- mcols(herv)$herv_id[exon_ol$herv_i]
exon_ol$gene_id <- exons$gene_id[exon_ol$exon_i]
exon_ol$gene_name <- exons$gene_name[exon_ol$exon_i]
exon_ol$overlap_bp <- width(pintersect(herv[exon_ol$herv_i], exons[exon_ol$exon_i], ignore.strand = TRUE))
exon_sum <- exon_ol %>%
group_by(herv_id) %>%
summarise(
n_exon_gene = n_distinct(gene_id),
exon_overlap_bp_max = max(overlap_bp),
exon_gene_list = paste(unique(gene_name), collapse = ";"),
.groups = "drop"
)
overlap_anno <- herv_base %>%
left_join(gene_sum, by = "herv_id") %>%
left_join(exon_sum, by = "herv_id") %>%
mutate(
across(c(n_gene_overlap, n_protein_coding_gene, same_strand_gene_n,
opposite_strand_gene_n, gene_overlap_bp_max, n_exon_gene,
exon_overlap_bp_max), ~replace_na(.x, 0)),
gene_overlap_frac_max = gene_overlap_bp_max / herv_len,
exon_overlap_frac_max = exon_overlap_bp_max / herv_len,
overlap_type = case_when(
n_exon_gene > 0 ~ "exonic",
n_gene_overlap > 0 ~ "intronic",
TRUE ~ "intergenic"
)
)
fwrite(overlap_anno, file.path(res_dir, "hERV_gene_overlap_annotation.csv"))
# 检验“重叠多是否表达高”
herv_assay <- "HERV"
expr_mat <- GetAssayData(seu, assay = herv_assay, slot = "data")
count_mat <- GetAssayData(seu, assay = herv_assay, slot = "counts")
expr_all <- data.frame(
herv_id_expr = rownames(expr_mat),
mean_expr_all = Matrix::rowMeans(expr_mat),
pct_expr_all = Matrix::rowMeans(count_mat > 0)
) %>%
filter(!grepl("no[-_]?feature", herv_id_expr, ignore.case = TRUE)) %>%
mutate(herv_id = gsub("-", "_", herv_id_expr))
ana_all <- expr_all %>%
left_join(overlap_anno, by = "herv_id") %>%
filter(!is.na(overlap_type))
stat_res <- data.frame(
test = c(
"mean_expr_vs_n_gene_overlap",
"mean_expr_vs_gene_overlap_frac",
"pct_expr_vs_n_gene_overlap",
"pct_expr_vs_gene_overlap_frac",
"mean_expr_by_overlap_type",
"pct_expr_by_overlap_type"
),
method = c("Spearman", "Spearman", "Spearman", "Spearman", "Kruskal-Wallis", "Kruskal-Wallis"),
p_value = c(
cor.test(ana_all$mean_expr_all, ana_all$n_gene_overlap, method = "spearman")$p.value,
cor.test(ana_all$mean_expr_all, ana_all$gene_overlap_frac_max, method = "spearman")$p.value,
cor.test(ana_all$pct_expr_all, ana_all$n_gene_overlap, method = "spearman")$p.value,
cor.test(ana_all$pct_expr_all, ana_all$gene_overlap_frac_max, method = "spearman")$p.value,
kruskal.test(mean_expr_all ~ overlap_type, data = ana_all)$p.value,
kruskal.test(pct_expr_all ~ overlap_type, data = ana_all)$p.value
),
estimate = c(
cor.test(ana_all$mean_expr_all, ana_all$n_gene_overlap, method = "spearman")$estimate,
cor.test(ana_all$mean_expr_all, ana_all$gene_overlap_frac_max, method = "spearman")$estimate,
cor.test(ana_all$pct_expr_all, ana_all$n_gene_overlap, method = "spearman")$estimate,
cor.test(ana_all$pct_expr_all, ana_all$gene_overlap_frac_max, method = "spearman")$estimate,
NA,
NA
)
)
fwrite(ana_all, file.path(res_dir, "hERV_overlap_expression_all_cells.csv"))
fwrite(stat_res, file.path(res_dir, "hERV_overlap_expression_stats_all_cells.csv"))
第一页表格:
- mean_expr_all:该hERV在所有细胞中的平均表达量
- pct_expr_all:该hERV在所有细胞中的检出比例
- n_gene_overlap:与该hERV有gene body重叠的GENCODE gene数量
- n_protein_coding_gene:重叠gene中protein-coding gene数量
- same/opposite_strand_gene_n:与hERV同/反义链重叠的gene数量
- gene_overlap_bp_max:与某个gene body的最大重叠长度
- gene_overlap_frac_max:gene_overlap_bp_max / herv_len
- gene_list:重叠gene列表
- n_exon_gene:与该hERV有exon重叠的gene数量(后面类似)
-
overlap_type:exonic/intronic/intergenic
gene_overlap_frac_max=1不代表hERV和外显子完全重叠。很多intronic hERV也会是1,因为它完整落在某个gene body内部
统计结果:前4行是Spearman秩相关cor.test(x, y, method = "spearman"),
- mean_expr_vs_n_gene_overlap:hERV平均表达量 vs 重叠gene数量
- mean_expr_vs_gene_overlap_frac:hERV平均表达量 vs 最大gene重叠比例
- pct_expr_vs_n_gene_overlap:hERV检出比例 vs 重叠gene数量
- pct_expr_vs_gene_overlap_frac:hERV检出比例 vs 最大gene重叠比例
- 后2行是Kruskal-Wallis检验
kruskal.test(a ~ b):比较exonic/intronic/intergenic三组的表达分布是否不同,只体现“三组是否存在总体差异”,不体现哪两组之间差异最大,所以estimate是NA
gene body和exon:
- gene body:从这个基因的起点到终点的整段区域,包含exon和intron
- exon:成熟转录本中保留下来的外显子区域,包括CDS和UTR
- 编码序列(Coding Sequence, CDS):编码蛋白质,决定氨基酸序列,位于mRNA中间
- 非翻译区(Untranslated Regions, UTR):非翻译区,位于mRNA两端,调控mRNA稳定性和翻译效率
- intron:在gene body内部,但不属于exon的区域
gene body: |-----------------------------|
exons: |---| |----| |-----|
introns: |------| |-------|
所以分类是:
- 如果hERV碰到了exon → exonic:最容易混入宿主gene成熟转录本或isoform信号
- 否则,如果hERV在gene body里面(hERV没有碰到任何exon) → intronic:可能是pre-mRNA、内含子保留、读穿转录,也可能是独立hERV转录
-
否则 → intergenic:很可能不是宿主gene转录本的一部分,但仍可能受附近增强子或启动子调控
gene body: |-----------------------------| exons: |---| |----| |-----| hERV: |------|这个hERV和第二个exon有重叠,也归为exonic。因为从转录解释上看,只要和exon重叠,就更可能混入宿主基因成熟转录本/isoform信号。也因此2024 NC–schERV+eQTL那篇文献会过滤掉“overlapping with gene exons”(HERVs that partially overlapped with gene exons were removed)
结论:
| overlap_type | 位点数 | 占比 |
|---|---|---|
| intergenic | 6155 | 43.34% |
| intronic | 5499 | 38.72% |
| exonic | 2549 | 17.95% |
| overlap_type | mean_expr_all中位数 | pct_expr_all中位数 |
|---|---|---|
| intergenic | 1.29e-05 | 1.45e-05 |
| intronic | 5.06e-05 | 5.80e-05 |
| exonic | 1.20e-04 | 1.84e-04 |
| strand_relation | 位点数 | mean_expr_all中位数 | pct_expr_all中位数 |
|---|---|---|---|
| none | 6155 | 1.29e-05 | 1.45e-05 |
| opposite_only | 3884 | 3.95e-05 | 4.59e-05 |
| both | 1374 | 1.13e-04 | 1.69e-04 |
| same_only | 2790 | 1.22e-04 | 1.59e-04 |
- 按中位数看,exonic hERV的平均表达大约是intergenic hERV的9倍左右,检出率大约是intergenic hERV的13倍左右:没有过滤gene exon重叠位点时,基因内/外显子重叠确实会明显抬高hERV表达信号(如果不过滤,确实容易混入宿主基因转录本或gene body转录活性带来的信号)
- rho大概在0.29–0.32之间:hERV表达/检出率和gene重叠程度有正相关,但不是强相关
- 在exonic hERV中,same-strand gene overlap位点的表达中位数也高于非same-strand位点;在intronic hERV中也是一样:same-strand重叠的hERV明显比opposite-strand重叠的hERV表达更高
2024 NC–schERV+eQTL这篇文献在过滤和外显子有重叠后,计算的是每个HERV的表达向量-最近gene的表达向量,也就是“HERV表达是否跟邻近gene表达同步变化”。上面做的是每个hERV位点的整体表达/检出率-这个hERV在基因组上与gene的重叠程度,也就是“表达量是否和基因组位置重叠有关”
再细分一下hERV和gene的重叠关系,然后按重叠关系分别计算hERV和gene表达相关性。比如说完全重合(且在同方向链上)的是不是相关性最高,部分重叠的其次,不重叠/在反义链上的最低?可以也效仿那篇文献的方法,看hERV与它最近gene表达的相关性?
| 分类 | 含义 | 解释 |
|---|---|---|
exonic_full_same |
hERV大部分被同链exon覆盖 | gene isoform/chimeric transcript候选 |
exonic_partial_same |
hERV部分与同链exon重叠 | - |
intronic_full_same |
hERV完整落在同链gene body内,但不与exon重叠 | 可能受gene转录环境影响,也可能是独立转录 |
intronic_partial_same |
hERV部分与同链gene body重叠 | - |
exonic_full_opposite / exonic_partial_opposite
|
与exon重叠但反义链 | 可能是独立/反义转录 |
intronic_full_opposite / intronic_partial_opposite
|
与gene body反义链重叠 | 可能是独立/反义转录 |
intergenic_nearest |
不与gene body重叠,只看最近gene | - |
# 重新分类
herv_df <- fread("/public/home/GENE_proc/wth/GSE174367/0530test/data/hERV_locus.bed", header = FALSE)
colnames(herv_df) <- c("chr", "start0", "end", "herv_id", "score", "strand")
herv <- GRanges(
seqnames = herv_df$chr,
ranges = IRanges(herv_df$start0 + 1, herv_df$end),
strand = herv_df$strand
)
mcols(herv)$herv_id <- herv_df$herv_id
mcols(herv)$herv_len <- width(herv)
gtf <- import("/public/home/wangtianhao/Desktop/STAR_ref/gencode.v49.annotation.gtf")
genes <- gtf[gtf$type == "gene"]
exons <- gtf[gtf$type == "exon"]
# 只看蛋白编码基因
genes <- genes[genes$gene_type == "protein_coding"]
exons <- exons[exons$gene_type == "protein_coding"]
std_chr <- paste0("chr", c(1:22, "X", "Y"))
herv <- keepSeqlevels(herv, intersect(seqlevels(herv), std_chr), pruning.mode = "coarse")
genes <- keepSeqlevels(genes, intersect(seqlevels(genes), std_chr), pruning.mode = "coarse")
exons <- keepSeqlevels(exons, intersect(seqlevels(exons), std_chr), pruning.mode = "coarse")
# 按gene合并exon,避免不同转录本exon重复计数
ex_by_gene <- split(exons, exons$gene_id)
ex_by_gene <- reduce(ex_by_gene)
exons_red <- unlist(ex_by_gene, use.names = FALSE)
exons_red$gene_id <- rep(names(ex_by_gene), elementNROWS(ex_by_gene))
# hERV-gene body overlap
gene_hits <- findOverlaps(herv, genes, ignore.strand = TRUE)
overlap_pair <- data.frame(
herv_i = queryHits(gene_hits),
gene_i = subjectHits(gene_hits)
)
if (nrow(overlap_pair) > 0) {
overlap_pair$herv_id <- mcols(herv)$herv_id[overlap_pair$herv_i]
overlap_pair$herv_len <- width(herv)[overlap_pair$herv_i]
overlap_pair$gene_id <- genes$gene_id[overlap_pair$gene_i]
overlap_pair$gene_name <- genes$gene_name[overlap_pair$gene_i]
overlap_pair$gene_type <- genes$gene_type[overlap_pair$gene_i]
overlap_pair$herv_strand <- as.character(strand(herv))[overlap_pair$herv_i]
overlap_pair$gene_strand <- as.character(strand(genes))[overlap_pair$gene_i]
overlap_pair$strand_relation <- ifelse(overlap_pair$herv_strand == overlap_pair$gene_strand, "same", "opposite")
overlap_pair$gene_overlap_bp <- width(pintersect(herv[overlap_pair$herv_i], genes[overlap_pair$gene_i], ignore.strand = TRUE))
overlap_pair$gene_overlap_frac <- overlap_pair$gene_overlap_bp / overlap_pair$herv_len
}
# hERV-exon overlap
exon_hits <- findOverlaps(herv, exons_red, ignore.strand = TRUE)
exon_pair <- data.frame(
herv_i = queryHits(exon_hits),
exon_i = subjectHits(exon_hits)
)
if (nrow(exon_pair) > 0) {
exon_pair$herv_id <- mcols(herv)$herv_id[exon_pair$herv_i]
exon_pair$gene_id <- exons_red$gene_id[exon_pair$exon_i]
exon_pair$exon_overlap_bp <- width(pintersect(herv[exon_pair$herv_i], exons_red[exon_pair$exon_i], ignore.strand = TRUE))
exon_sum <- exon_pair %>%
group_by(herv_id, gene_id) %>%
summarise(exon_overlap_bp = sum(exon_overlap_bp), .groups = "drop")
} else {
exon_sum <- data.frame(herv_id = character(), gene_id = character(), exon_overlap_bp = numeric())
}
# 给所有overlap pair分类
full_cutoff <- 0.8
overlap_pair <- overlap_pair %>%
left_join(exon_sum, by = c("herv_id", "gene_id")) %>%
mutate(
exon_overlap_bp = replace_na(exon_overlap_bp, 0),
exon_overlap_bp = pmin(exon_overlap_bp, herv_len),
exon_overlap_frac = exon_overlap_bp / herv_len,
loc_class = ifelse(exon_overlap_bp > 0, "exonic", "intronic"),
cover_class = case_when(
loc_class == "exonic" & exon_overlap_frac >= full_cutoff ~ "full",
loc_class == "exonic" ~ "partial",
loc_class == "intronic" & gene_overlap_frac >= full_cutoff ~ "full",
TRUE ~ "partial"
),
overlap_relation = paste(loc_class, cover_class, strand_relation, sep = "_"),
priority = case_when(
loc_class == "exonic" & strand_relation == "same" ~ 1,
loc_class == "intronic" & strand_relation == "same" ~ 2,
loc_class == "exonic" & strand_relation == "opposite" ~ 3,
loc_class == "intronic" & strand_relation == "opposite" ~ 4,
TRUE ~ 5
),
overlap_score = ifelse(loc_class == "exonic", exon_overlap_frac, gene_overlap_frac),
gene_choice = "overlap_gene"
)
best_overlap <- overlap_pair %>%
dplyr::arrange(herv_id, priority, dplyr::desc(overlap_score)) %>%
dplyr::group_by(herv_id) %>%
dplyr::filter(dplyr::row_number() == 1) %>%
dplyr::ungroup()
# 对没有任何gene body overlap的hERV,找最近TSS gene
overlap_herv <- unique(best_overlap$herv_id)
intergenic_herv <- herv[!mcols(herv)$herv_id %in% overlap_herv]
tss <- promoters(genes, upstream = 0, downstream = 1)
nearest_hit <- distanceToNearest(intergenic_herv, tss, ignore.strand = TRUE)
nearest_pair <- data.frame(
herv_i = queryHits(nearest_hit),
gene_i = subjectHits(nearest_hit),
nearest_gene_distance = mcols(nearest_hit)$distance
)
if (nrow(nearest_pair) > 0) {
nearest_pair$herv_id <- mcols(intergenic_herv)$herv_id[nearest_pair$herv_i]
nearest_pair$herv_len <- width(intergenic_herv)[nearest_pair$herv_i]
nearest_pair$gene_id <- genes$gene_id[nearest_pair$gene_i]
nearest_pair$gene_name <- genes$gene_name[nearest_pair$gene_i]
nearest_pair$gene_type <- genes$gene_type[nearest_pair$gene_i]
nearest_pair$herv_strand <- as.character(strand(intergenic_herv))[nearest_pair$herv_i]
nearest_pair$gene_strand <- as.character(strand(genes))[nearest_pair$gene_i]
nearest_pair$strand_relation <- ifelse(nearest_pair$herv_strand == nearest_pair$gene_strand, "same", "opposite")
nearest_pair$gene_overlap_bp <- 0
nearest_pair$gene_overlap_frac <- 0
nearest_pair$exon_overlap_bp <- 0
nearest_pair$exon_overlap_frac <- 0
nearest_pair$loc_class <- "intergenic"
nearest_pair$cover_class <- "none"
nearest_pair$overlap_relation <- "intergenic_nearest"
nearest_pair$gene_choice <- "nearest_gene"
}
# 合并成每个hERV唯一相关gene表
related_gene <- bind_rows(
best_overlap %>% select(
herv_id, herv_len, gene_id, gene_name, gene_type,
herv_strand, gene_strand, strand_relation,
gene_overlap_bp, gene_overlap_frac,
exon_overlap_bp, exon_overlap_frac,
loc_class, cover_class, overlap_relation, gene_choice
),
nearest_pair %>% select(
herv_id, herv_len, gene_id, gene_name, gene_type,
herv_strand, gene_strand, strand_relation,
gene_overlap_bp, gene_overlap_frac,
exon_overlap_bp, exon_overlap_frac,
loc_class, cover_class, overlap_relation, gene_choice,
nearest_gene_distance
)
)
related_gene <- related_gene %>%
mutate(
nearest_gene_distance = replace_na(nearest_gene_distance, 0),
herv_id_expr = gsub("_", "-", herv_id)
)
fwrite(related_gene, file.path(res_dir, "hERV_related_gene_annotation.csv"))
计算hERV-gene表达相关性:
related_gene <- read.csv(file.path(res_dir, "hERV_related_gene_annotation.csv"))
related_gene$herv_id_expr <- ifelse(
related_gene$herv_id %in% rownames(herv_mat),
related_gene$herv_id,
gsub("_", "-", related_gene$herv_id)
)
pair_use <- related_gene %>%
filter(herv_id_expr %in% rownames(herv_mat), gene_name %in% rownames(gene_mat)) %>%
distinct(herv_id, .keep_all = TRUE)
# 完整计算需5~6h,抽取1/3计算
# pair_use <- pair_use %>%
# dplyr::mutate(overlap_relation = as.character(overlap_relation)) %>%
# dplyr::group_by(overlap_relation) %>%
# dplyr::mutate(.rand = runif(dplyr::n())) %>%
# dplyr::arrange(overlap_relation, .rand) %>%
# dplyr::filter(dplyr::row_number() <= ceiling(dplyr::n() / 3)) %>%
# dplyr::ungroup() %>%
# dplyr::select(-.rand)
# print(table(pair_use_sub$overlap_relation))
corr_res <- lapply(seq_len(nrow(pair_use)), function(i) {
my_print(i, "/", nrow(pair_use))
h <- as.numeric(herv_mat[pair_use$herv_id_expr[i], ])
g <- as.numeric(gene_mat[pair_use$gene_name[i], ])
rho <- suppressWarnings(cor(h, g, method = "spearman"))
n <- length(h)
p <- ifelse(
is.na(rho) | abs(rho) >= 1,
NA,
2 * pt(-abs(rho * sqrt((n - 2) / (1 - rho^2))), df = n - 2)
)
data.frame(
herv_id = pair_use$herv_id[i],
herv_id_expr = pair_use$herv_id_expr[i],
gene_name = pair_use$gene_name[i],
gene_id = pair_use$gene_id[i],
gene_type = pair_use$gene_type[i],
gene_choice = pair_use$gene_choice[i],
loc_class = pair_use$loc_class[i],
cover_class = pair_use$cover_class[i],
strand_relation = pair_use$strand_relation[i],
overlap_relation = pair_use$overlap_relation[i],
gene_overlap_frac = pair_use$gene_overlap_frac[i],
exon_overlap_frac = pair_use$exon_overlap_frac[i],
nearest_gene_distance = pair_use$nearest_gene_distance[i],
rho = rho,
p_value = p,
herv_pct = Matrix::rowMeans(herv_count[pair_use$herv_id_expr[i], , drop = FALSE] > 0),
gene_pct = Matrix::rowMeans(gene_count[pair_use$gene_name[i], , drop = FALSE] > 0),
both_pct = mean(herv_count[pair_use$herv_id_expr[i], ] > 0 & gene_count[pair_use$gene_name[i], ] > 0),
stringsAsFactors = FALSE
)
}) %>%
bind_rows() %>%
mutate(p_adj = p.adjust(p_value, method = "BH"))
fwrite(corr_res, file.path(res_dir, "hERV_related_gene_expression_corr.csv"))
按重叠关系汇总相关性:
# 同链完全重叠、同链部分重叠、反义链、不重叠,它们的相关性是否依次下降
summary_by_relation <- corr_res %>%
group_by(overlap_relation) %>%
summarise(
n_pair = n(),
median_rho = median(rho, na.rm = TRUE),
mean_rho = mean(rho, na.rm = TRUE),
median_abs_rho = median(abs(rho), na.rm = TRUE),
q75_abs_rho = quantile(abs(rho), 0.75, na.rm = TRUE),
prop_rho_gt_0.1 = mean(rho > 0.1, na.rm = TRUE),
prop_rho_gt_0.2 = mean(rho > 0.2, na.rm = TRUE),
prop_abs_rho_gt_0.2 = mean(abs(rho) > 0.2, na.rm = TRUE),
median_herv_pct = median(herv_pct, na.rm = TRUE),
median_gene_pct = median(gene_pct, na.rm = TRUE),
median_both_pct = median(both_pct, na.rm = TRUE),
.groups = "drop"
) %>%
arrange(desc(median_rho))
# “全部”和“去掉exonic hERV后”的总相关性
summ_one <- function(df, label) {
df %>%
summarise(
group = label,
n_pair = n(),
median_rho = median(rho, na.rm = TRUE),
mean_rho = mean(rho, na.rm = TRUE),
median_abs_rho = median(abs(rho), na.rm = TRUE),
q75_abs_rho = quantile(abs(rho), 0.75, na.rm = TRUE),
prop_rho_gt_0.1 = mean(rho > 0.1, na.rm = TRUE),
prop_rho_gt_0.2 = mean(rho > 0.2, na.rm = TRUE),
prop_abs_rho_gt_0.2 = mean(abs(rho) > 0.2, na.rm = TRUE),
median_herv_pct = median(herv_pct, na.rm = TRUE),
median_gene_pct = median(gene_pct, na.rm = TRUE),
median_both_pct = median(both_pct, na.rm = TRUE)
)
}
summary_all_vs_no_exonic <- bind_rows(
summ_one(corr_res, "all_hERV"),
summ_one(corr_res %>% filter(loc_class != "exonic"), "no_exonic_hERV")
)
wb <- createWorkbook()
addWorksheet(wb, "summary_by_relation")
writeData(wb, "summary_by_relation", summary_by_relation)
addWorksheet(wb, "summary_all_vs_no_exonic")
writeData(wb, "summary_all_vs_no_exonic", summary_all_vs_no_exonic)
saveWorkbook(
wb,
file.path(res_dir, "hERV_related_gene_expression_corr_summary.xlsx"),
overwrite = TRUE
)
| 重叠关系 | n_pair | median_rho | mean_rho | rho>0.2比例 |
|---|---|---|---|---|
| exonic_full_same | 26 | 0.137 | 0.221 | 38.5% |
| exonic_partial_same | 188 | 0.119 | 0.175 | 33.0% |
| intronic_full_same | 620 | 0.0667 | 0.0964 | 11.2% |
| exonic_partial_opposite | 177 | 0.0127 | 0.0163 | 0% |
| intronic_full_opposite | 1817 | 0.0113 | 0.0163 | 0.06% |
| exonic_full_opposite | 22 | 0.0052 | 0.0081 | 0% |
| intergenic_nearest | 9047 | 0.0028 | 0.0065 | 0% |
-
同链exonic>同链intronic>反义链重叠≈intergenic最近gene:相关性不是单纯来自“离gene近”,而是明显依赖是否与gene同链、是否落在exon/gene body内部
| 来源 | rho>0.2数量 |
|---|---|
| intronic_full_same | 66 |
| exonic_partial_same | 61 |
| exonic_full_same | 10 |
| intronic_full_opposite | 1 |
| intergenic_nearest | 0 |
- 强相关pair几乎都来自同链重叠位点
| 分组 | n_pair | median_rho | mean_rho | rho>0.2比例 |
|---|---|---|---|---|
| all_hERV | 11897 | 0.0047 | 0.0175 | 1.40% |
| no_exonic_hERV | 11484 | 0.0044 | 0.0139 | 0.71% |
- 大多数pair是
intergenic_nearest,所以整体平均相关性非常低 - 去掉
exonic hERV以后,median几乎不变,但rho>0.2比例从1.40%降到0.71%:exonic hERV不会明显改变整体中位数,但会显著贡献强相关尾部信号
gene-hERV chimeric transcript (snRNA-seq)
基因和hERV嵌合转录本(gene-hERV chimeric transcript)是否在AD有特异表达的,这种转录本有潜力直接成为标志物/靶点
| 类型 | 含义 | 是否需要额外证据 |
|---|---|---|
| 真正gene-hERV嵌合转录本 | 同一条RNA里同时包含gene exon和hERV序列 | 必须要junction/split read或长读长证据 |
| LTR驱动的alternative promoter转录本 | HERV/LTR作为启动子,后面接宿主gene exon | 需要5’端证据、junction read或CAGE/long-read |
| 邻近调控型HERV-gene关系 | HERV作为enhancer/promoter-like元件调控gene | 需要ATAC/ChIP/EPI/eQTL等机制证据 |
使用A mouse-specific retrotransposon drives a conserved Cdk2ap1 isoform essential for development中的Retrotransposon:Gene Junction Reads Quantification方法。因为单细胞数据会非常稀疏,且该方法也不支持超大样本数,所以用pseudobulk的方法,得到junction×sample_celltype矩阵
- 该方法的关键输出:
- junction-by-sample矩阵:行是junction,列是sample,值是跨该junction的read数
- junction注释表:把junction和exon/RT重叠信息匹配起来,输出
info(chr:start:end)、gene_name、junction_type、max_te_perc、start_te_repName/start_te_info、end_te_repName/end_te_info等
- 我的pseudobulk输出:
-
all_junction_by_sample.csv:所有剪接junction在每个GSM中的read计数 -
gene_hERV_junction_info.csv:所有被注释为gene-hERV连接的junction,包括连接哪个gene、哪个hERV、哪一端是hERV、哪一端是gene exon -
gene_hERV_junction_by_sample.csv:只保留gene-hERV junction后的junction×sample矩阵 -
gene_hERV_junction_by_celltype_pseudobulk.csv:后续按celltype拆分BAM后得到的junction×sample_celltype矩阵
-
该pipeline共分为两条线:
- 表达/覆盖证据:BAM→BigWig→后续可视化
- 结构/junction证据:BAM→junction→gene/hERV边界匹配→嵌合转录本分类
1. STARsolo first-pass,重新生成SJ.out.tab:用的是常规参考基因组+GENCODE注释,STAR会在比对过程中发现剪接reads,比如“read的一部分比到exon A,另一部分比到exon B,中间跳过一段intron”,这些结果会记录在SJ.out.tab里,所以第一轮的作用是“从真实RNA-seq数据中发现新的splice junction”,特别是gene-hERV嵌合转录本里,很多junction不在GENCODE里,这种junction如果不在参考注释里,第一次比对可能能发现,但敏感性不一定最高
- 在STAR参数中加上了
--outSAMstrandField intronMotif,这样second-pass BAM里会有XS标签(后续可能用regtools -s XS)
module load miniconda3/base
conda activate STAR
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res/
genomeDir=/public/home/wangtianhao/Desktop/STAR_ref/hg38/
whitelist=/public/home/wangtianhao/Desktop/STAR_ref/whitelist/3M-february-2018.txt
mkdir -p ${res_dir}/star_1pass
run_star_1pass() {
dataset=$1
cdna_suffix=$2
bcumi_suffix=$3
umi_len=$4
workDir=/public/home/GENE_proc/wth/${dataset}
fqDir=${workDir}/fastq
mapFile=${fqDir}/srr2gsm.csv
outDir=${res_dir}/star_1pass/${dataset}
mkdir -p ${outDir}
dos2unix ${mapFile}
GSM_LIST=$(cut -d',' -f2 ${mapFile} | sort -u)
for GSM in ${GSM_LIST}; do
GSM=${GSM//$'\r'/}
echo "========== first-pass ${dataset} ${GSM} =========="
SRR_LIST=$(awk -F',' -v g="${GSM}" '$2==g{print $1}' ${mapFile})
cdna_files=$(echo "${SRR_LIST}" | sed "s|^|${fqDir}/|; s|$|_${cdna_suffix}.fastq.gz|" | paste -sd, -)
bcumi_files=$(echo "${SRR_LIST}" | sed "s|^|${fqDir}/|; s|$|_${bcumi_suffix}.fastq.gz|" | paste -sd, -)
for f in $(echo "${cdna_files}" | tr ',' ' ') $(echo "${bcumi_files}" | tr ',' ' '); do
if [[ ! -f "${f}" ]]; then
echo "[ERROR] Missing fastq: ${f}" >&2
exit 1
fi
done
STAR \
--runMode alignReads \
--runThreadN 16 \
--genomeDir ${genomeDir} \
--readFilesIn ${cdna_files} ${bcumi_files} \
--readFilesCommand zcat \
--outFileNamePrefix ${outDir}/${GSM} \
--soloType CB_UMI_Simple \
--soloCBstart 1 \
--soloCBlen 16 \
--soloUMIstart 17 \
--soloUMIlen ${umi_len} \
--soloBarcodeReadLength 0 \
--soloCBwhitelist ${whitelist} \
--soloFeatures GeneFull \
--clipAdapterType CellRanger4 \
--soloCellFilter EmptyDrops_CR \
--soloCBmatchWLtype 1MM_multi_Nbase_pseudocounts \
--soloUMIfiltering MultiGeneUMI_CR \
--soloUMIdedup 1MM_CR \
--outSAMtype BAM SortedByCoordinate \
--outSAMattributes NH HI nM AS CR UR CB UB GX GN sS sQ sM \
--outSAMstrandField intronMotif \
--outSAMunmapped Within \
--limitOutSJcollapsed 5000000 \
--outFilterScoreMin 30 \
--outFilterMultimapNmax 500 \
--outFilterMultimapScoreRange 5 \
--alignSJoverhangMin 8 \
--alignIntronMin 50 \
--alignIntronMax 500000
done
}
run_star_1pass GSE157827 2 1 10
run_star_1pass GSE174367 3 2 12
mv ${res_dir}/star_1pass/GSE157827/*SJ.out.tab ${res_dir}/star_1pass/
mv ${res_dir}/star_1pass/GSE174367/*SJ.out.tab ${res_dir}/star_1pass/
2. 汇总first-pass junction,构建updated STAR index。先从first-pass结果里选可信junction,再把这些junction加入STAR索引用于second-pass
原pipeline在此步:从first-pass的SJ.out.tab里筛canonical splicing junction,要求至少3条reads支持,再用这些junction更新STAR索引。但我这里执行后得到了211w条junction,而STAR默认最多插入1000000条,可能是因为样本太多+snRNA-seq junction较碎+STAR参数放宽。因此把筛选条件改严格一些:
- 只保留主染色体
- canonical junction:$5>0
- novel junction:$6==0,因为GENCODE注释junction已经由–sjdbGTFfile提供了,不需要重复插入
- 每个junction至少在2个样本出现
- 全部样本累计unique reads≥5或10
- 最大overhang≥12或20
module load miniconda3/base
conda activate STAR
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res/
genome_fa=/public/home/wangtianhao/Desktop/STAR_ref/GRCh38.p14.genome.fa
gtf=/public/home/wangtianhao/Desktop/STAR_ref/gencode.v49.annotation.gtf
out_index=${res_dir}/STAR_ref_2pass_hERV_junc
mkdir -p ${res_dir}/SJ_filtered
mkdir -p ${out_index}
# cat ${res_dir}/star_1pass/GSE157827/*SJ.out.tab ${res_dir}/star_1pass/GSE174367/*SJ.out.tab \
# | awk 'BEGIN{FS=OFS="\t"}
# {
# if($5 > 0 && $7 > 2 && $6 == 0 && $4 == 1) print $1,$2,$3,"+";
# else if($5 > 0 && $7 > 2 && $6 == 0 && $4 == 2) print $1,$2,$3,"-";
# }' \
# | sort -u > ${res_dir}/SJ_filtered/SJ.filtered.tab
cat ${res_dir}/star_1pass/*SJ.out.tab \
| awk 'BEGIN{FS=OFS="\t"}
$1 ~ /^chr([1-9]|1[0-9]|2[0-2]|X|Y)$/ && $5 > 0 && $6 == 0 && $7 >= 1 && $9 >= 12 && ($4 == 1 || $4 == 2) {
strand=($4==1?"+":"-")
key=$1 FS $2 FS $3 FS strand
count[key] += 1
reads[key] += $7
if($9 > overhang[key]) overhang[key] = $9
}
END{
for(k in count) {
if(count[k] >= 2 && reads[k] >= 5 && overhang[k] >= 12) print k
}
}' \
| sort -u > ${res_dir}/SJ_filtered/SJ.filtered.tab
STAR \
--runThreadN 16 \
--runMode genomeGenerate \
--genomeDir ${out_index} \
--genomeFastaFiles ${genome_fa} \
--sjdbGTFfile ${gtf} \
--sjdbFileChrStartEnd ${res_dir}/SJ_filtered/SJ.filtered.tab \
--sjdbOverhang 99
3. STARsolo second-pass,生成最终BAM和SJ.out.tab:第二次比对时,STAR就相当于“提前知道这些junction可能存在”,所以更容易把跨junction reads正确比上,提高junction read的回收率
module load miniconda3/base
conda activate STAR
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res/
genomeDir=${res_dir}/STAR_ref_2pass_hERV_junc
whitelist=/public/home/wangtianhao/Desktop/STAR_ref/whitelist/3M-february-2018.txt
mkdir -p ${res_dir}/star_2pass
run_star_2pass() {
dataset=$1
cdna_suffix=$2
bcumi_suffix=$3
umi_len=$4
workDir=/public/home/GENE_proc/wth/${dataset}
fqDir=${workDir}/fastq
mapFile=${fqDir}/srr2gsm.csv
outDir=${res_dir}/star_2pass/${dataset}
mkdir -p ${outDir}
dos2unix ${mapFile}
GSM_LIST=$(cut -d',' -f2 ${mapFile} | sort -u)
for GSM in ${GSM_LIST}; do
GSM=${GSM//$'\r'/}
echo "========== second-pass ${dataset} ${GSM} =========="
SRR_LIST=$(awk -F',' -v g="${GSM}" '$2==g{print $1}' ${mapFile})
cdna_files=$(echo "${SRR_LIST}" | sed "s|^|${fqDir}/|; s|$|_${cdna_suffix}.fastq.gz|" | paste -sd, -)
bcumi_files=$(echo "${SRR_LIST}" | sed "s|^|${fqDir}/|; s|$|_${bcumi_suffix}.fastq.gz|" | paste -sd, -)
for f in $(echo "${cdna_files}" | tr ',' ' ') $(echo "${bcumi_files}" | tr ',' ' '); do
if [[ ! -f "${f}" ]]; then
echo "[ERROR] Missing fastq: ${f}" >&2
exit 1
fi
done
STAR \
--runMode alignReads \
--runThreadN 16 \
--genomeDir ${genomeDir} \
--readFilesIn ${cdna_files} ${bcumi_files} \
--readFilesCommand zcat \
--outFileNamePrefix ${outDir}/${GSM} \
--soloType CB_UMI_Simple \
--soloCBstart 1 \
--soloCBlen 16 \
--soloUMIstart 17 \
--soloUMIlen ${umi_len} \
--soloBarcodeReadLength 0 \
--soloCBwhitelist ${whitelist} \
--soloFeatures GeneFull \
--clipAdapterType CellRanger4 \
--soloCellFilter EmptyDrops_CR \
--soloCBmatchWLtype 1MM_multi_Nbase_pseudocounts \
--soloUMIfiltering MultiGeneUMI_CR \
--soloUMIdedup 1MM_CR \
--outSAMtype BAM SortedByCoordinate \
--outSAMattributes NH HI nM AS CR UR CB UB GX GN sS sQ sM \
--outSAMstrandField intronMotif \
--outSAMunmapped Within \
--limitOutSJcollapsed 5000000 \
--outFilterScoreMin 30 \
--outFilterMultimapNmax 500 \
--outFilterMultimapScoreRange 5 \
--alignSJoverhangMin 8 \
--alignIntronMin 50 \
--alignIntronMax 500000
samtools index -@8 ${outDir}/${GSM}Aligned.sortedByCoord.out.bam
done
}
run_star_2pass GSE157827 2 1 10
run_star_2pass GSE174367 3 2 12
4. 从second-pass的SJ.out.tab生成junction bed
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res/
mkdir -p ${res_dir}/sample_junctions_from_STAR
for dataset in GSE157827 GSE174367; do
for sj in ${res_dir}/star_2pass/${dataset}/*SJ.out.tab; do
[[ -f ${sj} ]] || continue
GSM=$(basename ${sj} SJ.out.tab)
echo "[SJ] ${dataset} ${GSM}"
awk -v sample="${dataset}_${GSM}" 'BEGIN{OFS="\t"}
{
strand="."
if($4==1) strand="+"
if($4==2) strand="-"
if($5>0 && $7>=1 && $9>=8 && strand!=".") {
start0=$2-1
end=$3
jid=$1":"start0":"end":"strand
print $1,start0,end,jid,$7,strand
}
}' ${sj} > ${res_dir}/sample_junctions_from_STAR/${dataset}_${GSM}.junc.bed
done
done
5. 生成Normalized BigWig:主要用于后续IGV看候选位点,不会直接参与junction矩阵和统计
-
--minMappingQuality 255对应STAR唯一比对reads(文档方法),对于hERV这种重复区域,这会牺牲敏感性,但可视化更保守 - BigWig:基因组覆盖度轨道文件,不是用来直接识别嵌合转录本的,而是用来看
- 某个基因/hERV区域有没有reads覆盖
- AD样本和NC样本覆盖度是否不同
- junction附近是否有连续表达信号
- hERV区域是否像一个exon一样被覆盖
但BigWig本身只能告诉你“这个区域有多少reads覆盖”,不能证明这些reads来自同一条转录本。真正证明gene和hERV连接的是junction read
conda activate hERV_coverage
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res/
mkdir -p ${res_dir}/bigwig
find ${res_dir}/star_2pass -name "*Aligned.sortedByCoord.out.bam" | while read bam; do
dataset=$(basename $(dirname ${bam}))
GSM=$(basename ${bam} Aligned.sortedByCoord.out.bam)
echo "[bigwig] ${dataset} ${GSM}"
bamCoverage \
--bam ${bam} \
-o ${res_dir}/bigwig/${dataset}_${GSM}.uniq.CPM.bw \
--minMappingQuality 255 \
--normalizeUsing CPM \
--binSize 10 \
-p 4
done
6. StringTie2逐样本组装,TACO合并:生成assembly.gtf,帮助判断hERV是参与了alternative promoter、internal exon还是terminator
- 10x3’snRNA-seq不太适合完整转录本组装,所以这一步结果要谨慎看,主要服务于结构解释和IGV展示
-
StringTie:根据RNA-seq reads组装转录本结构,利用exon覆盖度、junction reads、read pair信息去推断可能存在的转录本模型
但对于3’的10xsnRNA-seq,由于reads集中在转录本3’端且单个细胞覆盖度低,对完整转录本组装并不理想
-
TACO:StringTie是每个样本单独组装,TACO的作用是把这些单样本组装结果合并成一个统一的转录本集合,这样后面的
overlap_exon_with_te.r就可以用一个统一的assembly.gtf来分析assembly.gtf:这个样本里推断出了哪些gene/transcript/exon,它们在基因组上的坐标是什么、由哪些exon组成、表达支持大概多强GL000008.2 taco transcript 156382 159928 1000 - . tss_id "TSS5"; locus_id "L4"; abs_frac "1.00000"; rel_frac "1.00000"; expr "0.074"; transcript_id "TU7"; gene_id "G3"; GL000008.2 taco exon 156382 157581 1000 - . tss_id "TSS5"; locus_id "L4"; transcript_id "TU7"; gene_id "G3"; GL000008.2 taco exon 158045 159928 1000 - . tss_id "TSS5"; locus_id "L4"; transcript_id "TU7"; gene_id "G3";表示:TACO在GL000008.2:156382-159928负链上,合并出一个转录本TU7,它属于gene/locus G3,这个转录本的TSS编号是TSS5,在该locus中的丰度信息/表达水平记录在abs_frac/rel_frac/expr中,这个转录本由两个exon组成——GL000008.2(156382-157581)、GL000008.2(chr1:158045-159928)
- TU7不是GENCODE原始transcript ID,它是TACO合并后重新命名的转录本单元,表示这个assembly.gtf中的第7号transcription unit/transcript model
- 同理G3也是TACO给这个locus/gene unit分配的ID,可以理解成TACO认为这些转录本属于同一个转录单元/基因区域G3
- TSS5是TACO推断的转录起始位点编号,同一个locus_id下可能有多个TSS
-
TU2 abs_frac "0.46376"; TU1 abs_frac "0.53624"表示TACO认为在这个局部区域里,TU1和TU2都存在,TU1占比略高 - 相对比例可以理解成相对于同一TSS或同一局部转录单元中最强转录本的比例。
TU1 rel_frac "1.00000"; TU2 rel_frac "0.86484"表示TU1是该组里相对最强的模型,TU2强度约为TU1的86% -
GL000008.2是参考基因组里的alternative contig(重复序列和特殊组装区域),后面也有正常的chrx开头的
为什么要合并:StringTie是根据reads覆盖和junction reads推断转录本结构,不同样本测序深度、细胞组成、表达强度不同,看到的reads范围也不同。真实转录本是exon1(1000-1500)+exon2(3000-5000),但样本A在exon1的5’端覆盖不够,StringTie可能只看到1020-1500,样本B覆盖更完整,可能看到980-1500,样本C测序深度更低,可能只组装出3000-5000
module load miniconda3/base
conda create -n stringtie_taco stringtie taco -y
conda activate stringtie_taco
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res/
gtf=/public/home/wangtianhao/Desktop/STAR_ref/gencode.v49.annotation.gtf
mkdir -p ${res_dir}/stringtie
find ${res_dir}/star_2pass -name "*Aligned.sortedByCoord.out.bam" | while read bam; do
dataset=$(basename $(dirname ${bam}))
GSM=$(basename ${bam} Aligned.sortedByCoord.out.bam)
echo "[stringtie] ${dataset} ${GSM}"
stringtie ${bam} \
-p 4 \
-G ${gtf} \
-j 2 \
-s 5 \
-f 0.05 \
-c 2 \
-o ${res_dir}/stringtie/${dataset}_${GSM}.stringtie.gtf
done
ls ${res_dir}/stringtie/*.stringtie.gtf > ${res_dir}/stringtie/gtf_to_merge.txt
taco_run \
-o ${res_dir}/stringtie/merged_gtf \
-p 4 \
${res_dir}/stringtie/gtf_to_merge.txt
7. 收集splicing junction并合并矩阵:从BAM里找含N的CIGAR reads
-
CIGAR(Compact Idiosyncratic Gapped Alignment Report):使用数字+字母组合表示reads的比对情况 更多关于bam比对结果的说明
比如一个read的CIGAR是
50M1000N50M,含义是:前50bp比到一个exon,跳过1000bp intron,后50bp比到另一个exon。此时这个read就是一个spliced read,可以支持一个junction -
作者脚本做的是:把BAM
- 只保留唯一比对reads
- 只保留含N的spliced reads
- 转成BED12
- 从BED block结构推断junction
- 统计每个junction有多少reads支持
conda create -n te_junc \
-c conda-forge -c bioconda \
python=3.8 perl samtools parallel \
r-base=4.2 r-optparse r-data.table r-magrittr r-reshape \
bioconductor-genomicranges bioconductor-iranges bioconductor-s4vectors bioconductor-genomeinfodb \
-y
从second-pass BAM提取junction:
conda activate te_junc
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res/
script_dir=/public/home/GENE_proc/wth/GSE174367/0530test/new_code/
mkdir -p ${res_dir}/junc_raw
find ${res_dir}/star_2pass -name "*Aligned.sortedByCoord.out.bam" > ${res_dir}/bam.list
parallel -j 4 '
bam={}
dataset=$(basename $(dirname ${bam}))
GSM=$(basename ${bam} Aligned.sortedByCoord.out.bam)
echo "[junc] ${dataset} ${GSM}"
'${script_dir}'/bam2junc_uniq_fixed.sh \
${bam} \
'${res_dir}'/junc_raw/${dataset}_${GSM}.junc
' :::: ${res_dir}/bam.list
合并junction矩阵:和gene表达矩阵类似,但行不是gene,而是junction;值不是gene表达量,而是跨这个junction的read数
find ${res_dir}/junc_raw -name "*.junc" | sort > ${res_dir}/junc_file_list.txt
Rscript ${script_dir}/merge_junction_files_fixed.r \
-j ${res_dir}/junc_file_list.txt \
-p 4 \
-n ${res_dir}/all_samples
# 检查输出
ls -lh ${res_dir}/all_samples_junc_combined.txt # 这个文件就是后续核心的junction×sample矩阵
head ${res_dir}/all_samples_junc_combined.txt
8. 把hERV_locus.bed转成脚本需要的repeat文件
这里把start加1,是为了从BED的0-based坐标转成R/GRanges更适合的1-based坐标。对junction边界匹配来说,原脚本有±20bp左右的窗口,1bp差异一般不改变结论,但最好从源头统一
herv_bed=/public/home/GENE_proc/wth/GSE174367/0530test/data/hERV_locus.bed
herv_clean=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res/hERV_locus.cleaned.txt
awk 'BEGIN{OFS="\t"; print "chr","start","end","strand","repName","repClass","repFamily"}
{
id=$4
gsub("_","-",id)
strand="*"
if(NF>=6 && ($6=="+" || $6=="-")) strand=$6
print $1,$2+1,$3,strand,id,"LTR","hERV"
}' ${herv_bed} > ${herv_clean}
head ${herv_clean}
9. 找哪些exon和hERV重叠:把转录本结构和hERV位置联系起来,使用基因gtf注释+转录本结构assembly.gtf+hERV位点注释,分析哪些exon和hERV区域重叠、exon的起点或终点是否落在hERV附近、这个exon有多少比例来自hERV
conda activate te_junc
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res/
script_dir=/public/home/GENE_proc/wth/GSE174367/0530test/new_code/
mkdir -p ${res_dir}/AD_hERV
Rscript ${script_dir}/overlap_exon_with_te_fixed.r \
-g /public/home/wangtianhao/Desktop/STAR_ref/gencode.v49.annotation.gtf \
-t ${res_dir}/stringtie/merged_gtf/assembly.gtf \
-r /public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res/hERV_locus.cleaned.txt \
-p 4 \
-n ${res_dir}/AD_hERV
这个文件会给每个exon加上:te_perc、start_edge_with_te、end_edge_with_te,后面Step10会用这些信息判断hERV是在junction的哪一端、属于promoter/internal/terminator哪一类
10. 识别gene-hERV junction并分类-:真正把junction和hERV/gene结构连起来,使用junction×sample矩阵+exon-hERV重叠结果+hERV注释,判断junction的一端是不是gene exon、junction的另一端是不是hERV-derived exon
conda activate te_junc
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res/
script_dir=/public/home/GENE_proc/wth/GSE174367/0530test/new_code/
Rscript ${script_dir}/overlap_junction_with_exon_fixed.r \
-e ${res_dir}/AD_hERV_exon_collection.txt \
-j ${res_dir}/all_samples_junc_combined.txt \
-r /public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res/hERV_locus.cleaned.txt \
-p 4 \
-n ${res_dir}/AD_hERV
预览 | 下载,注:这个表是按样本(group)聚合的,没有做细胞类型的划分
汇总AD/Control聚合结果:
x <- as.data.table(read_xlsx(file.path(res_dir, "AD_hERV_processed_junctions.xlsx")))
sample_cols <- grep("^GSE", names(x), value = TRUE)
x[, total_reads := rowSums(.SD), .SDcols = sample_cols]
x[, n_sample_detected := rowSums(.SD > 0), .SDcols = sample_cols]
x_filt <- x[
total_reads >= 10 &
n_sample_detected >= 2 &
max_te_perc >= 5
]
# 排除Y染色体结果
x_filt[, chr := sub(":.*", "", info)]
x_filt[, is_chrY := chr == "chrY"]
# 构造junction_id
x_filt[, herv_name := fifelse(
te_side == "start", start_te_repName,
fifelse(te_side == "end", end_te_repName,
paste(start_te_repName, end_te_repName, sep = "|"))
)]
x_filt[, junction_id := paste(info, gene_name, herv_name, junction_type, sep = "|")]
anno_cols <- setdiff(names(x_filt), sample_cols)
long <- melt(
x_filt,
id.vars = anno_cols,
measure.vars = sample_cols,
variable.name = "sample_uid",
value.name = "count"
)
# 读取样本信息
meta <- fread("C:/Users/17185/Desktop/hERV_AD_snRNA-seq_analyze/metadata/gsm_info.csv")
if (!"sample_uid" %in% names(meta)) {
meta[, sample_uid := paste(dataset, GSM, sep = "_")]
}
long <- merge(
long,
meta[, .(sample_uid, dataset, GSM, group, donor_uid)],
by = "sample_uid",
all.x = TRUE
)
# 用所有junction矩阵作为测序深度归一化分母
all_junc_path <- file.path(res_dir, "all_samples_junc_combined.txt")
all_junc <- fread(all_junc_path)
depth <- data.table(
sample_uid = sample_cols,
junc_depth = colSums(all_junc[, ..sample_cols])
)
long <- merge(long, depth, by = "sample_uid", all.x = TRUE)
long[, cpm := count / junc_depth * 1e6]
# 按group聚合
group_summary <- long[, .(
n_sample = .N,
n_detected = sum(count > 0),
detect_rate = mean(count > 0),
mean_count = mean(count),
sum_count = sum(count),
mean_cpm = mean(cpm),
median_cpm = median(cpm)
), by = .(
junction_id, info, gene_name, herv_name, junction_type,
te_side, max_te_perc, chr, is_chrY, group
)]
group_wide <- dcast(
group_summary,
junction_id + info + gene_name + herv_name + junction_type +
te_side + max_te_perc + chr + is_chrY ~ group,
value.var = c("mean_cpm", "detect_rate", "sum_count", "n_detected"),
fill = 0
)
group_wide[, log2FC_AD_vs_Control := log2((mean_cpm_AD + 0.1) / (mean_cpm_NC + 0.1))]
group_wide[, delta_detect_rate := detect_rate_AD - detect_rate_NC]
# 统计检验:检测率Fisher + CPM Wilcoxon
test_res <- long[, {
g <- group
detected <- count > 0
p_fisher <- tryCatch({
fisher.test(table(g, detected))$p.value
}, error = function(e) NA_real_)
p_wilcox <- tryCatch({
wilcox.test(cpm ~ g)$p.value
}, error = function(e) NA_real_)
.(
fisher_p = p_fisher,
wilcox_p = p_wilcox
)
}, by = junction_id]
test_res[, fisher_q := p.adjust(fisher_p, method = "BH")]
test_res[, wilcox_q := p.adjust(wilcox_p, method = "BH")]
group_wide <- merge(group_wide, test_res, by = "junction_id", all.x = TRUE)
fwrite(
group_wide,
file.path(res_dir, "AD_hERV_junction_group_summary.tsv"),
sep = "\t"
)
| 类型 | 数量 | 解释 |
|---|---|---|
| internal | 449 | hERV在转录本中间,作为internal exon或内部剪接位点 |
| terminator | 194 | hERV可能参与末端外显子/3’端/3’UTR样结构 |
| promoter_tss | 64 | 最可能是“HERV提供5’端exon / alternative promoter” |
| promoter | 7 | 也属于5’端候选 |
| internal/terminator | 137 | 同一个junction在不同转录本模型中可解释为internal或terminal |
| internal/promoter_tss | 78 | |
| 其它复合类型 | 35 |
这些分类是怎么分出来的:
- 先给每个exon定位:
- (转录方向上的)第1个exon → first_exon
- 中间exon → internal_exon
- 最后1个exon → last_exon
- 再看junction连接到了哪个exon:如果一个junction的一端是hERV重叠的exon,另一端是gene exon,就可能被识别为gene-hERV junction
- 最后根据hERV所在exon的位置分类
- promoter:hERV所在的一端对应的是转录本第一个exon——这个hERV-derived exon位于转录本5’端附近,结构上像是hERV提供了一个新的5’端外显子
HERV-exon → gene exon2 → gene exon3 → ...- promoter_tss:hERV不仅在first exon里,而且覆盖了first exon的边界,也就是更靠近转录起始端
HERV边界/TSS附近 → HERV-derived first exon → downstream gene exon- internal:hERV-derived exon位于转录本中间,不是第一个exon,也不是最后一个exon
gene exon1 → gene exon2 → HERV-exon → gene exon4- terminator:hERV-derived exon位于转录本最后一个exon
gene exon1 → gene exon2 → gene exon3 → HERV-exon / 3'UTR
表格中的te_side:这个junction的哪一端被判定为hERV/TE相关端
- start/end是基因组坐标上的junction起点/终点(坐标靠前的就是start端),不是转录方向上的5’/3’
| te_side | 含义 | 简单理解 |
|---|---|---|
start |
junction的start端落在hERV/TE相关exon或hERV边界附近 | junction的左端是hERV端 |
end |
junction的end端落在hERV/TE相关exon或hERV边界附近 | junction的右端是hERV端 |
both |
start端和end端都检测到hERV/TE重叠 | 两端都和hERV有关,解释时要更谨慎 |
在AD-NC差异方面,没有达到FDR显著的。因为junction read非常稀疏,而且样本数较少
| junction | gene | hERV | 类型 | AD mean CPM | NC mean CPM | AD-NC检出率差 | 备注 |
|---|---|---|---|---|---|---|---|
| chr2:71380565:71388622 | ENSG00000075292.21 | HML5-2p13.2 | internal | 0.91 | 0.22 | +0.414 | 最强AD倾向之一,Fisher/Wilcox nominal显著 |
| chr2:71388723:71393393 | ENSG00000075292.21 | HML5-2p13.2 | internal | 1.27 | 0.45 | +0.325 | 与上一个位点同一区域,可能是同一转录结构的一部分 |
| chr16:2663407:2671594 | ENSG00000260565.10 | HML4-16p13.3 | internal/terminator | 0.37 | 0.08 | +0.317 | 可能涉及末端或内部exon |
| chr6:97373914:97423468 | ENSG00000271860.10 | ERVL-6q16.1c | internal | 0.20 | 0.03 | +0.317 | AD检出率差较明显 |
| chr12:42441429:42442640 | ENSG00000134283.19 | HML5-12q12a | internal/terminator | 0.48 | 0.06 | +0.286 | 适合看IGV |
| chr19:41479284:41495301 | ENSG00000267107.10 | ERV316A3-19q13.2 | internal | 7.79 | 2.49 | +0.123 | CPM高,可能更稳 |
| chr19:41495427:41500583 | ENSG00000267107.10 | ERV316A3-19q13.2 | internal/terminator | 5.86 | 2.21 | +0.107 | 与上一个相邻,可能代表同一isoform区域 |
-
chr2:HML5-2p13.2–ENSG00000075292.21和chr19:ERV316A3-19q13.2–ENSG00000267107.10不是单个孤立junction,而是同一区域有多个相邻junction同时表现出AD升高趋势
promoter相关(5’端候选):
| junction | gene | hERV | 类型 | AD mean CPM | NC mean CPM | 备注 |
|---|---|---|---|---|---|---|
| chr20:46433884:46436461 | ENSG00000310436.1 | ERV316A3-20q13.12 | internal/promoter_tss | 0.25 | 0 | AD中有reads,NC近乎无 |
| chr22:36134024:36135090 | ENSG00000304491.1 | ERVL-22q12.3b | promoter_tss | 0.23 | 0 | 典型promoter_tss候选 |
| chr19:53865728:53869753 | 多个ENSG | ERVLE-19q13.42 | promoter_tss | 0.32 | 0.03 | 需解决多gene注释 |
| chr22:37190928:37195380 | ENSG00000133466.15 | MER41-22q12.3e | internal/promoter | 2.79 | 1.01 | CPM较高 |
| chr13:83650443:83815676 | ENSG00000286446.2 | MER101-13q31.1a | promoter_tss | 0.16 | 0.006 | AD倾向较强 |
- 注意:promoter_tss/promoter只能说明“结构上像5’端HERV exon”,不能单独证明RNA真的从HERV处起始
terminator相关(3’端/3’UTR候选):
| junction | gene | hERV | 类型 | AD mean CPM | NC mean CPM | 备注 |
|---|---|---|---|---|---|---|
| chr12:42441429:42442640 | ENSG00000134283.19 | HML5-12q12a | internal/terminator | 0.48 | 0.06 | AD倾向明显 |
| chr16:2663407:2671594 | ENSG00000260565.10 | HML4-16p13.3 | internal/terminator | 0.37 | 0.08 | 检出率差明显 |
| chr19:41495427:41500583 | ENSG00000267107.10 | ERV316A3-19q13.2 | internal/terminator | 5.86 | 2.21 | read数高 |
| chr2:95123826:95148884 | ENSG00000289685.2 / ENSG00000144026.15 | ERVLB4-2q11.1a | internal/terminator | 0.58 | 0.20 | 可能是3’端候选 |
| chr19:57513151:57516053 | 多个ENSG | HML6-19q13.43a | terminator | 0.76 | 0.35 | 需看多gene注释 |
- 注:上述两个表格中有的检测出的count数和count分布在样本数都比较第,比如
chr20:46433884:46436461只在2个AD样本中有90个count
鉴定哪些转录本使用了HERV作为alternative promote:
- 上述方法可以回答“哪些gene转录本中出现了HERV/LTR来源的5’端exon,并且该HERV/LTR通过splice junction连接到下游gene exon”
- alternative promoter至少包含两个层面的证据:
- 转录结构证据:HERV/LTR位于转录本5’端,并且通过splice junction连接到下游gene exon
- 转录起始证据:RNA真的从HERV/LTR内部或附近起始,而不是HERV只是被包含进了一个更长的转录本
- 10x数据主要捕获3’端,虽然BAM里仍可能有部分跨junction reads,但它不是专门为TSS/isoform结构设计的,容易漏检TSS,也容易把pre-mRNA、未完全剪接RNA或低水平背景junction误认为isoform
假设一个gene有4个外显子:
Promoter_gene → exon1 → exon2 → exon3 → exon4
如果HERV作为alternative promoter,结构应该是:
HERV/LTR promoter → HERV-derived exon → exon2 → exon3 → exon4
即RNA从HERV/LTR开始,然后剪接到下游gvene exon。这里HERV/LTR替代了原来的gene promoter和canonical exon1。但如果HERV只是internal exon,结构可能是:
Promoter_gene → exon1 → exon2 → HERV-derived exon → exon3 → exon4
这种情况下也会有HERV-gene junction,但HERV不是promoter,只是被纳入了转录本中间
具体来说,当前数据可以做的是junction证据/isoform结构证据:
- HERV/LTR位于gene上游或gene内部靠近5’端,并且与gene在同一条链上
- junction一端落在HERV/LTR区域,另一端落在下游gene exon
- HERV/LTR端是转录本最5’端exon或第一外显子附近,而不是gene中间某个internal exon
- junction_type被pipeline标注为promoter_tss或promoter
- 该junction在多个样本中有read支持
- 下游gene exon表达存在,但canonical 5’ exon不一定同步升高(如果HERV替代了原来的gene promoter,那么这个isoform会使用下游exon,但不使用canonical first exon),这说明它可能是一个不同起始位点的isoform
- HERV/LTR区域在对应细胞类型中开放
但更直接的TSS证据需要5’RACE/CAGE/RAMPAGE/PRO-cap/长读长等方法显示5’端RNA从HERV/LTR起始
感觉如果想要看“哪些转录本使用了HERV作为alternative promote”,也许不一定要找AD显著升高的?可以等bulk RNA-seq的结果,因为snRNA的结果还是有些稀疏
gene-hERV chimeric transcript (bulk RNA-seq)
1. 整理从SRA上直接下载的metadata
import pandas as pd
from pathlib import Path
fq_dir = Path("/public/home/GENE_proc/wth/GSE174367/fastq_bulkRNA")
res_dir = Path("/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res_bulk")
meta = pd.read_csv(fq_dir / "metadata.csv")
bulk = meta[
(meta["Assay Type"] == "RNA-Seq") &
(meta["isolation"] == "total RNA isolation")
].copy()
bulk["SRR"] = bulk["Run"].astype(str)
bulk["GSM"] = bulk["GEO_Accession (exp)"].astype(str)
bulk["group"] = bulk["diagnosis"].astype(str)
cols = ["SRR", "GSM", "group", "brain_region", "AGE", "sex", "pmi", "LibraryLayout", "LibrarySelection", "LibrarySource"]
cols = [x for x in cols if x in bulk.columns]
bulk = bulk[cols].drop_duplicates()
bulk.to_csv(res_dir / "metadata" / "bulk_sample_table.tsv", sep="\t", index=False)
bulk[["GSM", "group", "brain_region", "AGE", "sex", "pmi"]].drop_duplicates().to_csv(
res_dir / "metadata" / "bulk_gsm_metadata.tsv", sep="\t", index=False
)
bulk["GSM"].drop_duplicates().sort_values().to_csv(
res_dir / "metadata" / "bulk_gsm_list.txt", index=False, header=False
)
bulk["SRR"].drop_duplicates().sort_values().to_csv(
res_dir / "metadata" / "bulk_srr_list.txt", index=False, header=False
)
2. STAR first-pass
module load miniconda3/base
conda activate STAR
fq_dir=/public/home/GENE_proc/wth/GSE174367/fastq_bulkRNA
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res_bulk
genomeDir=/public/home/wangtianhao/Desktop/STAR_ref/hg38
sample_table=${res_dir}/metadata/bulk_sample_table.tsv
gsm_list=${res_dir}/metadata/bulk_gsm_list.txt
mkdir -p ${res_dir}/star_1pass ${res_dir}/logs
while read GSM; do
echo "========== STAR first-pass ${GSM} =========="
out_prefix=${res_dir}/star_1pass/${GSM}
if [[ -s ${out_prefix}SJ.out.tab && -s ${out_prefix}Aligned.sortedByCoord.out.bam ]]; then
echo "[skip] first-pass exists: ${GSM}"
continue
fi
SRR_LIST=$(awk -F'\t' -v gsm="${GSM}" 'NR>1 && $2==gsm{print $1}' ${sample_table})
r1_files=$(echo "${SRR_LIST}" | sed "s|^|${fq_dir}/|; s|$|_1.fastq.gz|" | paste -sd, -)
r2_files=$(echo "${SRR_LIST}" | sed "s|^|${fq_dir}/|; s|$|_2.fastq.gz|" | paste -sd, -)
for f in $(echo "${r1_files},${r2_files}" | tr ',' ' '); do
if [[ ! -f "${f}" ]]; then
echo "[ERROR] Missing fastq: ${f}" >&2
exit 1
fi
done
STAR \
--runMode alignReads \
--runThreadN 16 \
--genomeDir ${genomeDir} \
--readFilesIn ${r1_files} ${r2_files} \
--readFilesCommand zcat \
--outFileNamePrefix ${out_prefix} \
--outSAMtype BAM SortedByCoordinate \
--outSAMstrandField intronMotif \
--outSAMattributes NH HI AS nM \
--outSAMunmapped Within \
--limitOutSJcollapsed 5000000 \
--limitBAMsortRAM 50000000000 \
--outFilterScoreMin 30 \
--outFilterMultimapNmax 500 \
--outFilterMultimapScoreRange 5 \
--alignSJoverhangMin 8 \
--alignIntronMin 50 \
--alignIntronMax 500000
samtools index -@ 16 ${out_prefix}Aligned.sortedByCoord.out.bam
done < ${gsm_list}
cd ${res_dir}
mkdir -p temp
mv star_1pass/*SJ.out.tab temp
rm -rf star_1pass
mv temp star_1pass
3. 汇总first-pass junction并筛选SJ:因为这里试了一下SJ数量没有超过100w,所以直接用作者的方法
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res_bulk
mkdir -p ${res_dir}/SJ_filtered
cat ${res_dir}/star_1pass/*SJ.out.tab | awk '{FS="\t";OFS="\t"} {if ($5 > 0 && $7 > 2 && $6==0 && $4==1) print $1, $2, $3, "+"; else if ($5 > 0 && $7 > 2 && $6==0 && $4==2) print $1, $2, $3, "-";}' | sort | uniq > ${res_dir}/SJ_filtered/SJ.filtered.tab
wc -l ${res_dir}/SJ_filtered/SJ.filtered.tab
head ${res_dir}/SJ_filtered/SJ.filtered.tab
4. STAR second-pass
module load miniconda3/base
conda activate STAR
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res_bulk
genome_fa=/public/home/wangtianhao/Desktop/STAR_ref/GRCh38.p14.genome.fa
gtf=/public/home/wangtianhao/Desktop/STAR_ref/gencode.v49.annotation.gtf
out_index=${res_dir}/STAR_ref_2pass
mkdir -p ${out_index}
STAR \
--runThreadN 16 \
--runMode genomeGenerate \
--genomeDir ${out_index} \
--genomeFastaFiles ${genome_fa} \
--sjdbGTFfile ${gtf} \
--sjdbFileChrStartEnd ${res_dir}/SJ_filtered/SJ.filtered.tab \
--sjdbOverhang 99 \
--limitSjdbInsertNsj 3000000
fq_dir=/public/home/GENE_proc/wth/GSE174367/fastq_bulkRNA
genomeDir=${res_dir}/STAR_ref_2pass
sample_table=${res_dir}/metadata/bulk_sample_table.tsv
gsm_list=${res_dir}/metadata/bulk_gsm_list.txt
mkdir -p ${res_dir}/star_2pass ${res_dir}/logs
while read GSM; do
echo "========== STAR second-pass ${GSM} =========="
out_prefix=${res_dir}/star_2pass/${GSM}
if [[ -s ${out_prefix}SJ.out.tab && -s ${out_prefix}Aligned.sortedByCoord.out.bam ]]; then
echo "[skip] second-pass exists: ${GSM}"
continue
fi
SRR_LIST=$(awk -F'\t' -v gsm="${GSM}" 'NR>1 && $2==gsm{print $1}' ${sample_table})
r1_files=$(echo "${SRR_LIST}" | sed "s|^|${fq_dir}/|; s|$|_1.fastq.gz|" | paste -sd, -)
r2_files=$(echo "${SRR_LIST}" | sed "s|^|${fq_dir}/|; s|$|_2.fastq.gz|" | paste -sd, -)
for f in $(echo "${r1_files},${r2_files}" | tr ',' ' '); do
if [[ ! -f "${f}" ]]; then
echo "[ERROR] Missing fastq: ${f}" >&2
exit 1
fi
done
STAR \
--runMode alignReads \
--runThreadN 16 \
--genomeDir ${genomeDir} \
--readFilesIn ${r1_files} ${r2_files} \
--readFilesCommand zcat \
--outFileNamePrefix ${out_prefix} \
--outSAMtype BAM SortedByCoordinate \
--outSAMstrandField intronMotif \
--outSAMattributes NH HI AS nM \
--outSAMunmapped Within \
--limitOutSJcollapsed 5000000 \
--limitBAMsortRAM 50000000000 \
--outFilterScoreMin 30 \
--outFilterMultimapNmax 500 \
--outFilterMultimapScoreRange 5 \
--alignSJoverhangMin 8 \
--alignIntronMin 50 \
--alignIntronMax 500000
samtools index -@ 8 ${out_prefix}Aligned.sortedByCoord.out.bam
done < ${gsm_list}
rm -rf ${res_dir}/star_2pass/*.out
rm -rf ${res_dir}/star_2pass/*_STARtmp
5. 生成BigWig
module load miniconda3/base
conda activate hERV_coverage
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res_bulk
gsm_list=${res_dir}/metadata/bulk_gsm_list.txt
mkdir -p ${res_dir}/bigwig
while read GSM; do
echo "========== bigWig ${GSM} =========="
bam=${res_dir}/star_2pass/${GSM}Aligned.sortedByCoord.out.bam
out_bw=${res_dir}/bigwig/${GSM}.uniq.CPM.bw
if [[ -s ${out_bw} ]]; then
echo "[skip] bigwig exists: ${GSM}"
continue
fi
bamCoverage \
--bam ${bam} \
-o ${out_bw} \
--minMappingQuality 255 \
--normalizeUsing CPM \
--binSize 10 \
-p 8
done < ${gsm_list}
6. StringTie+TACO
module load miniconda3/base
conda activate stringtie_taco
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res_bulk
gtf=/public/home/wangtianhao/Desktop/STAR_ref/gencode.v49.annotation.gtf
gsm_list=${res_dir}/metadata/bulk_gsm_list.txt
mkdir -p ${res_dir}/stringtie
while read GSM; do
echo "========== StringTie ${GSM} =========="
bam=${res_dir}/star_2pass/${GSM}Aligned.sortedByCoord.out.bam
out_gtf=${res_dir}/stringtie/${GSM}.stringtie.gtf
if [[ -s ${out_gtf} ]]; then
echo "[skip] stringtie exists: ${GSM}"
continue
fi
stringtie ${bam} \
-p 8 \
-G ${gtf} \
-j 2 \
-s 5 \
-f 0.05 \
-c 2 \
-o ${out_gtf}
done < ${gsm_list}
ls ${res_dir}/stringtie/*.stringtie.gtf > ${res_dir}/stringtie/gtf_to_merge.txt
taco_run \
-o ${res_dir}/stringtie/merged_gtf \
-p 8 \
${res_dir}/stringtie/gtf_to_merge.txt
ls -lh ${res_dir}/stringtie/merged_gtf/assembly.gtf
7. 从second-pass BAM提取junction并合并
module load miniconda3/base
conda activate te_junc
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res_bulk
script_dir=${res_dir}/scripts
gsm_list=${res_dir}/metadata/bulk_gsm_list.txt
mkdir -p ${res_dir}/junctions
find ${res_dir}/star_2pass -name "*Aligned.sortedByCoord.out.bam" | sort > ${res_dir}/bam.list
parallel -j 4 --halt soon,fail=1 '
bam={}
GSM=$(basename "${bam}" Aligned.sortedByCoord.out.bam)
out="'${res_dir}'/junctions/${GSM}.junc"
if [[ -s "${out}" ]]; then
echo "[skip] ${GSM}"
exit 0
fi
echo "[junc] ${GSM}"
bash "'${script_dir}'/bam2junc_uniq_fixed.sh" \
"${bam}" \
"${out}"
' :::: ${res_dir}/bam.list
find ${res_dir}/junctions -name "*.junc" | sort > ${res_dir}/junctions/junc_file_list.txt
Rscript ${script_dir}/merge_junction_files_fixed.r \
-j ${res_dir}/junctions/junc_file_list.txt \
-p 1 \
-n ${res_dir}/junctions/bulk_all_samples
head /public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res_bulk/junctions/bulk_all_samples_junc_combined.txt
head /public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res_bulk/stringtie/merged_gtf/assembly.gtf
8. 找哪些exon和hERV重叠+识别gene-hERV junction并分类
module load miniconda3/base
conda activate te_junc
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res_bulk
script_dir=${res_dir}/scripts
gtf=/public/home/wangtianhao/Desktop/STAR_ref/gencode.v49.annotation.gtf
assembly_gtf=${res_dir}/stringtie/merged_gtf/assembly.gtf
herv_clean=/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res/hERV_locus.cleaned.txt
Rscript ${script_dir}/overlap_exon_with_te_fixed.r \
-g ${gtf} \
-t ${assembly_gtf} \
-r ${herv_clean} \
-p 4 \
-n ${res_dir}/bulk_hERV
# ls -lh ${res_dir}/bulk_hERV_exon_collection.txt
# head ${res_dir}/bulk_hERV_exon_collection.txt
Rscript ${script_dir}/overlap_junction_with_exon_fixed.r \
-e ${res_dir}/bulk_hERV_exon_collection.txt \
-j ${res_dir}/junctions/bulk_all_samples_junc_combined.txt \
-r ${herv_clean} \
-p 4 \
-n ${res_dir}/bulk_hERV
# ls -lh ${res_dir}/bulk_hERV_processed_junctions.txt
# head ${res_dir}/bulk_hERV_processed_junctions.txt
9. 汇总AD/Control聚合结果
library(data.table)
res_dir <- "/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res_bulk"
processed_path <- file.path(res_dir, "bulk_hERV_processed_junctions.txt")
all_junc_path <- file.path(res_dir, "junctions/bulk_all_samples_junc_combined.txt")
meta_path <- file.path(res_dir, "metadata/bulk_gsm_metadata.tsv")
x <- fread(processed_path)
all_junc <- fread(all_junc_path)
meta <- fread(meta_path)
sample_cols <- grep("GSM[0-9]+", names(x), value = TRUE)
sample_map <- data.table(sample_col = sample_cols)
sample_map[, GSM := sub(".*(GSM[0-9]+).*", "\\1", sample_col)]
sample_map <- sample_map[GSM %in% meta$GSM]
sample_cols <- sample_map$sample_col
x[, chr := sub(":.*", "", info)]
x[, total_reads := rowSums(.SD), .SDcols = sample_cols]
x[, n_sample_detected := rowSums(.SD > 0), .SDcols = sample_cols]
x_filt <- x[
total_reads >= 10 &
n_sample_detected >= 2 &
max_te_perc >= 5 &
chr != "chrY"
]
x_filt[, hERV_name := fifelse(
te_side == "start", start_te_repName,
fifelse(te_side == "end", end_te_repName, paste(start_te_repName, end_te_repName, sep = "/"))
)]
x_filt[, hERV_family := fifelse(
te_side == "start", start_te_repFamily,
fifelse(te_side == "end", end_te_repFamily, paste(start_te_repFamily, end_te_repFamily, sep = "/"))
)]
x_filt[, hERV_class := fifelse(
te_side == "start", start_te_repClass,
fifelse(te_side == "end", end_te_repClass, paste(start_te_repClass, end_te_repClass, sep = "/"))
)]
info_cols <- c(
"info", "chr", "gene_name", "junction_type", "te_side", "max_te_perc",
"hERV_name", "hERV_family", "hERV_class",
"start_te_repName", "start_te_repFamily", "start_te_repClass", "start_te_info",
"end_te_repName", "end_te_repFamily", "end_te_repClass", "end_te_info",
"total_reads", "n_sample_detected"
)
info_cols <- intersect(info_cols, names(x_filt))
gene_herv_info <- unique(x_filt[, ..info_cols])
fwrite(gene_herv_info, file.path(res_dir, "bulk_gene_hERV_junction_info.tsv"), sep = "\t")
mat <- x_filt[, c(info_cols, sample_cols), with = FALSE]
fwrite(mat, file.path(res_dir, "bulk_gene_hERV_junction_by_sample.tsv"), sep = "\t")
junc_sample_cols <- grep("GSM[0-9]+", names(all_junc), value = TRUE)
junc_sample_map <- data.table(sample_col = junc_sample_cols)
junc_sample_map[, GSM := sub(".*(GSM[0-9]+).*", "\\1", sample_col)]
junc_sample_map <- junc_sample_map[GSM %in% meta$GSM]
depth_long <- melt(
all_junc[, c("info", junc_sample_map$sample_col), with = FALSE],
id.vars = "info",
variable.name = "sample_col",
value.name = "count"
)
depth <- depth_long[, .(junc_depth = sum(count)), by = sample_col]
long <- melt(
x_filt[, c(info_cols, sample_cols), with = FALSE],
id.vars = info_cols,
variable.name = "sample_col",
value.name = "count"
)
long[, sample_col := as.character(sample_col)]
long <- merge(long, sample_map, by = "sample_col", all.x = TRUE)
long <- merge(long, meta, by = "GSM", all.x = TRUE)
long <- merge(long, depth, by = "sample_col", all.x = TRUE)
long[, cpm := count / junc_depth * 1e6]
fwrite(long, file.path(res_dir, "bulk_gene_hERV_junction_long.tsv"), sep = "\t")
summarize_group <- function(dt, by_extra, out_file) {
by_cols <- c(
"info", "chr", "gene_name", "junction_type", "te_side", "max_te_perc",
"hERV_name", "hERV_family", "hERV_class",
by_extra, "group"
)
by_cols <- intersect(by_cols, names(dt))
s <- dt[, .(
n_sample = .N,
n_detected = sum(count > 0),
detect_rate = mean(count > 0),
mean_count = mean(count),
sum_count = sum(count),
mean_cpm = mean(cpm),
median_cpm = median(cpm)
), by = by_cols]
id_cols <- setdiff(by_cols, "group")
w <- dcast(
s,
as.formula(paste(paste(id_cols, collapse = " + "), "~ group")),
value.var = c("n_sample", "n_detected", "detect_rate", "mean_count", "sum_count", "mean_cpm", "median_cpm"),
fill = 0
)
if (all(c("mean_cpm_AD", "mean_cpm_Control") %in% names(w))) {
w[, log2FC_AD_vs_Control := log2((mean_cpm_AD + 0.1) / (mean_cpm_Control + 0.1))]
}
if (all(c("detect_rate_AD", "detect_rate_Control") %in% names(w))) {
w[, delta_detect_rate_AD_vs_Control := detect_rate_AD - detect_rate_Control]
}
test_res <- dt[, {
fisher_p <- tryCatch(fisher.test(table(group, count > 0))$p.value, error = function(e) NA_real_)
wilcox_p <- tryCatch(wilcox.test(cpm ~ group)$p.value, error = function(e) NA_real_)
.(fisher_p = fisher_p, wilcox_p = wilcox_p)
}, by = id_cols]
test_res[, fisher_q := p.adjust(fisher_p, method = "BH")]
test_res[, wilcox_q := p.adjust(wilcox_p, method = "BH")]
w <- merge(w, test_res, by = id_cols, all.x = TRUE)
fwrite(w, file.path(res_dir, out_file), sep = "\t")
}
summarize_group(long, character(0), "bulk_gene_hERV_junction_group_summary.tsv")
summarize_group(long, "brain_region", "bulk_gene_hERV_junction_region_group_summary.tsv")
| 文件 | 行数 | 说明 |
|---|---|---|
bulk_gene_hERV_junction_info |
990条junction | 每条junction的注释信息 |
bulk_gene_hERV_junction_by_sample |
990条junction×191个样本 | 每条junction在各样本中的数量 |
bulk_gene_hERV_junction_group_summary |
990条junction | 每条junction按AD/Control汇总 |
bulk_gene_hERV_junction_region_group_summary |
1980行 | 每条junction按2个脑区(FC/CB)汇总 |
| junction_type | 数量 |
|---|---|
| internal | 410 |
| terminator | 186 |
| internal/terminator | 178 |
| internal/promoter_tss | 95 |
| promoter_tss | 59 |
| 其它混合类型 | 少量 |
- 不过统计结果整体不强,最小p值有比较小的,但校正后不显著
汇总
| 种类 | 数量 |
|---|---|
| bulk RNA-seq中的gene-hERV junction数 | 990 |
| snRNA-seq中的gene-hERV junction数 | 964 |
| 按info+gene+hERV合并后的非冗余junction数 | 1379 |
| 两个结果都出现 | 575 |
| 只在bulk结果表出现 | 415 |
| 只在snRNA结果表出现 | 389 |
| bulk和snRNA中AD侧CPM和检测率均更高 | 58 |
| bulk和snRNA中AD侧CPM和检测率均更低 | 104 |
| bulk RNA-seq没有BH校正后显著 | 61 |
| snRNA-seq没有BH校正后显著 | 45 |
| bulk RNA-seq经BH校正后显著 | 0 |
| snRNA-seq经BH校正后显著 | 0 |
| Structure class | bulk count | snRNA count |
|---|---|---|
| promoter_candidate | 216 | 184 |
| terminator_candidate | 364 | 331 |
| internal_candidate | 410 | 449 |
用pyGenomeTracks看一下候选位点
把已有样本级BigWig按AD/NC平均:
module load miniconda3/base
conda activate hERV_coverage
threads=${THREADS:-8}
sn_res_dir=${SN_RES_DIR:-/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res}
bulk_res_dir=${BULK_RES_DIR:-/public/home/GENE_proc/wth/GSE174367/0530test/chimeric_res_bulk}
sn_meta=${SN_META:-/public/home/GENE_proc/wth/metadata/gsm_info.csv}
bulk_meta=${BULK_META:-${bulk_res_dir}/metadata/bulk_gsm_metadata.tsv}
out_dir=${OUT_DIR:-/public/home/GENE_proc/wth/GSE174367/0530test/gene_hERV_browser}
mkdir -p ${out_dir}/lists
# snRNA:根据gsm_info.csv整理sample_uid和group
python - <<PY
import csv
sn_meta = "${sn_meta}"
out_tsv = "${out_dir}/lists/snRNA_sample_group.tsv"
with open(sn_meta, newline="") as f:
sample = f.read(4096)
f.seek(0)
dialect = csv.Sniffer().sniff(sample, delimiters=",\t")
reader = csv.DictReader(f, dialect=dialect)
rows = []
for r in reader:
dataset = r.get("dataset", "").strip()
gsm = r.get("GSM", "").strip()
group = r.get("group", "").strip()
if dataset and gsm and group in ["AD", "NC"]:
rows.append((dataset, gsm, group))
with open(out_tsv, "w") as f:
f.write("dataset\tGSM\tgroup\n")
for dataset, gsm, group in rows:
f.write(f"{dataset}\t{gsm}\t{group}\n")
PY
sn_meta=${SN_META:-${out_dir}/lists/snRNA_sample_group.tsv}
mkdir -p ${out_dir}/bigwig_group_avg
# snRNA
for group in AD NC; do
sn_list=${out_dir}/lists/snRNA.${group}.bw.list
awk -F'\t' -v g="${group}" 'NR>1 && $3==g {print $1"\t"$2}' ${sn_meta} \
| while IFS=$'\t' read -r dataset GSM; do
bw=${sn_res_dir}/bigwig/${dataset}_${GSM}.uniq.CPM.bw
[[ -s ${bw} ]] && echo ${bw}
done > ${sn_list}
n=$(wc -l < ${sn_list})
echo "[snRNA ${group}] BigWig number: ${n}"
if [[ ${n} -eq 0 ]]; then
echo "[ERROR] No BigWig found for snRNA ${group}" >&2
exit 1
fi
out_bw=${out_dir}/bigwig_group_avg/snRNA.${group}.meanCPM.bw
if [[ ${n} -eq 1 ]]; then
cp $(cat ${sn_list}) ${out_bw}
else
bigwigAverage \
-b $(cat ${sn_list}) \
-o ${out_bw} \
-p ${threads}
fi
done
# bulk
for group in AD Control; do
bulk_list=${out_dir}/lists/bulk.${group}.bw.list
awk -F'\t' -v g="${group}" 'NR>1 && $2==g {print $1}' ${bulk_meta} \
| while read GSM; do
bw=${bulk_res_dir}/bigwig/${GSM}.uniq.CPM.bw
[[ -s ${bw} ]] && echo ${bw}
done > ${bulk_list}
n=$(wc -l < ${bulk_list})
echo "[bulk ${group}] BigWig number: ${n}"
if [[ ${n} -eq 0 ]]; then
echo "[ERROR] No BigWig found for bulk ${group}" >&2
exit 1
fi
out_bw=${out_dir}/bigwig_group_avg/bulk.${group}.meanCPM.bw
if [[ ${n} -eq 1 ]]; then
cp $(cat ${bulk_list}) ${out_bw}
else
bigwigAverage \
-b $(cat ${bulk_list}) \
-o ${out_bw} \
-p ${threads}
fi
done
pyGenomeTracks画图:
junction=$1
gene_name=$2
herv_name=$3
junction_type=$4
range=${5:-30000}
module load miniconda3/base
root_dir=${ROOT_DIR:-/public/home/GENE_proc/wth/GSE174367/0530test/gene_hERV_browser}
bw_dir=${BW_DIR:-${root_dir}/bigwig_group_avg}
gtf=${GTF:-/public/home/wangtianhao/Desktop/STAR_ref/gencode.v49.annotation.gtf}
herv_bed=${HERV_BED:-/public/home/GENE_proc/wth/GSE174367/0530test/data/hERV_locus.bed}
out_root=${OUT_ROOT:-${root_dir}/track_plots}
bulk_ad_bw=${BULK_AD_BW:-${bw_dir}/bulk.AD.meanCPM.bw}
bulk_nc_bw=${BULK_NC_BW:-${bw_dir}/bulk.Control.meanCPM.bw}
sn_ad_bw=${SNRNA_AD_BW:-${bw_dir}/snRNA.AD.meanCPM.bw}
sn_nc_bw=${SNRNA_NC_BW:-${bw_dir}/snRNA.NC.meanCPM.bw}
threads=${THREADS:-4}
ymax_bulk=${YMAX_BULK:-5}
ymax_sn=${YMAX_SNRNA:-5}
diff_ymax=${DIFF_YMAX:-3}
chr=$(echo ${junction} | cut -d':' -f1)
junc_start=$(echo ${junction} | cut -d':' -f2)
junc_end=$(echo ${junction} | cut -d':' -f3)
region_start=$((junc_start - range))
if [[ ${region_start} -lt 0 ]]; then region_start=0; fi
region_end=$((junc_end + range))
region=${chr}:${region_start}-${region_end}
safe_id=$(echo "${junction}_${gene_name}_${herv_name}_${junction_type}" | sed 's#[/:| ]#_#g')
plot_dir=${out_root}/${safe_id}_${range}bp
mkdir -p ${plot_dir}
for f in "${bulk_ad_bw}" "${bulk_nc_bw}" "${sn_ad_bw}" "${sn_nc_bw}" "${gtf}" "${herv_bed}"; do
if [[ ! -s ${f} ]]; then
echo "[ERROR] File not found: ${f}" >&2
exit 1
fi
done
herv_norm=$(echo "${herv_name}" | sed 's#|#/#g' | cut -d'/' -f1 | sed 's/-/_/g')
awk -v h="${herv_norm}" -v c="${chr}" 'BEGIN{OFS="\t"} $1==c && $4==h {print}' ${herv_bed} \
> ${plot_dir}/candidate_hERV.body.bed
if [[ ! -s ${plot_dir}/candidate_hERV.body.bed ]]; then
echo "[WARN] Exact hERV name not found in hERV_locus.bed: ${herv_name} -> ${herv_norm}" >&2
echo "[WARN] Only junction and nearby hERV tracks will be drawn." >&2
fi
# candidate junction弧线,BEDPE格式
left_end=$((junc_start + 1))
right_start=$((junc_end - 1))
echo -e "${chr}\t${junc_start}\t${left_end}\t${chr}\t${right_start}\t${junc_end}\t${gene_name}|${herv_name}|${junction_type}\t1000\t+" \
> ${plot_dir}/candidate_junction.bedpe
# junction两个边界
echo -e "${chr}\t${junc_start}\t${left_end}\tjunction_left\t0\t+" > ${plot_dir}/junction_boundary.bed
echo -e "${chr}\t${right_start}\t${junc_end}\tjunction_right\t0\t+" >> ${plot_dir}/junction_boundary.bed
# 附近所有hERV
awk -v c="${chr}" -v s=${region_start} -v e=${region_end} 'BEGIN{OFS="\t"} $1==c && $2<e && $3>s {print}' \
${herv_bed} > ${plot_dir}/nearby_hERV.bed
awk -v h="${herv_norm}" '$4!=h' ${plot_dir}/nearby_hERV.bed > ${plot_dir}/nearby_hERV.no_candidate.bed
# gene span
awk -v c="${chr}" -v s=${region_start} -v e=${region_end} 'BEGIN{OFS="\t"}
$1==c && $4<=e && $5>=s && $3=="gene" {
name=$0
if(name ~ /gene_name "/){sub(/.*gene_name "/, "", name); sub(/".*/, "", name)} else {name=$9}
print $1, $4-1, $5, name, 0, $7
}' ${gtf} > ${plot_dir}/genes.span.bed
# exon
awk -v c="${chr}" -v s=${region_start} -v e=${region_end} 'BEGIN{OFS="\t"}
$1==c && $4<=e && $5>=s && $3=="exon" {
name=$0
if(name ~ /gene_name "/){sub(/.*gene_name "/, "", name); sub(/".*/, "", name)} else {name=$9}
print $1, $4-1, $5, name, 0, $7
}' ${gtf} > ${plot_dir}/genes.exon.bed
python - <<PY
import re
gtf = "${gtf}"
chr_ = "${chr}"
region_start = int("${region_start}")
region_end = int("${region_end}")
target = "${gene_name}"
plot_dir = "${plot_dir}"
target_nover = target.split(".")[0]
def get_attr(attr, key):
m = re.search(key + r' "([^"]+)"', attr)
return m.group(1) if m else ""
span = []
exon = []
with open(gtf) as f:
for line in f:
if line.startswith("#"):
continue
x = line.rstrip("\n").split("\t")
if len(x) < 9:
continue
if x[0] != chr_:
continue
feature = x[2]
start = int(x[3]) - 1
end = int(x[4])
strand = x[6]
attr = x[8]
if end < region_start or start > region_end:
continue
if feature not in ["gene", "exon"]:
continue
gene_id = get_attr(attr, "gene_id")
gene_name = get_attr(attr, "gene_name")
gene_id_nover = gene_id.split(".")[0]
label = gene_name if gene_name else gene_id
row = [x[0], str(start), str(end), label, "0", strand, gene_id, gene_name, gene_id_nover]
if feature == "gene":
span.append(row)
elif feature == "exon":
exon.append(row)
def is_target(row):
gene_id = row[6]
gene_symbol = row[7]
gene_id_nover = row[8]
return target in [gene_id, gene_symbol, gene_id_nover] or target_nover in [gene_id, gene_symbol, gene_id_nover]
with open(f"{plot_dir}/genes.span.bed", "w") as f:
for r in span:
f.write("\t".join(r[:6]) + "\n")
with open(f"{plot_dir}/genes.exon.bed", "w") as f:
for r in exon:
f.write("\t".join(r[:6]) + "\n")
with open(f"{plot_dir}/target_gene.span.bed", "w") as f:
for r in span:
if is_target(r):
f.write("\t".join(r[:6]) + "\n")
with open(f"{plot_dir}/other_genes.span.bed", "w") as f:
for r in span:
if not is_target(r):
f.write("\t".join(r[:6]) + "\n")
with open(f"{plot_dir}/target_gene.exon.bed", "w") as f:
for r in exon:
if is_target(r):
f.write("\t".join(r[:6]) + "\n")
with open(f"{plot_dir}/other_genes.exon.bed", "w") as f:
for r in exon:
if not is_target(r):
f.write("\t".join(r[:6]) + "\n")
PY
conda activate hERV_coverage
bigwigCompare \
-b1 ${bulk_ad_bw} \
-b2 ${bulk_nc_bw} \
--operation subtract \
--binSize 10 \
--region ${chr}:${region_start}:${region_end} \
-p ${threads} \
-o ${plot_dir}/bulk.AD_minus_NC.bw
bigwigCompare \
-b1 ${sn_ad_bw} \
-b2 ${sn_nc_bw} \
--operation subtract \
--binSize 10 \
--region ${chr}:${region_start}:${region_end} \
-p ${threads} \
-o ${plot_dir}/snRNA.AD_minus_NC.bw
cat > ${plot_dir}/${safe_id}.tracks.ini <<EOF
[x-axis]
where = top
[bulk AD]
file = ${bulk_ad_bw}
title = bulk AD CPM
height = 2
color = #d73027
min_value = 0
max_value = ${ymax_bulk}
number_of_bins = 900
nans_to_zeros = true
[bulk NC]
file = ${bulk_nc_bw}
title = bulk NC CPM
height = 2
color = #4575b4
min_value = 0
max_value = ${ymax_bulk}
number_of_bins = 900
nans_to_zeros = true
[bulk AD-NC]
file = ${plot_dir}/bulk.AD_minus_NC.bw
title = bulk AD-NC
height = 1.6
color = #555555
min_value = -${diff_ymax}
max_value = ${diff_ymax}
number_of_bins = 900
nans_to_zeros = true
grid = true
[snRNA AD]
file = ${sn_ad_bw}
title = snRNA AD CPM
height = 2
color = #d73027
min_value = 0
max_value = ${ymax_sn}
number_of_bins = 900
nans_to_zeros = true
[snRNA NC]
file = ${sn_nc_bw}
title = snRNA NC CPM
height = 2
color = #4575b4
min_value = 0
max_value = ${ymax_sn}
number_of_bins = 900
nans_to_zeros = true
[snRNA AD-NC]
file = ${plot_dir}/snRNA.AD_minus_NC.bw
title = snRNA AD-NC
height = 1.6
color = #555555
min_value = -${diff_ymax}
max_value = ${diff_ymax}
number_of_bins = 900
nans_to_zeros = true
grid = true
[candidate junction]
file = ${plot_dir}/candidate_junction.bedpe
title = candidate junction
height = 1.4
file_type = links
links_type = arcs
line_width = 2
color = #7b3294
[junction boundary]
file = ${plot_dir}/junction_boundary.bed
title = junction boundary
height = 0.5
display = collapsed
labels = false
color = #7b3294
[candidate hERV]
file = ${plot_dir}/candidate_hERV.body.bed
title = candidate hERV
height = 0.7
display = collapsed
labels = true
color = red
[nearby hERVs]
file = ${plot_dir}/nearby_hERV.no_candidate.bed
title = nearby hERVs
height = 1.2
display = stacked
labels = true
color = grey
[other genes]
file = ${plot_dir}/other_genes.span.bed
title = other genes
height = 0.6
display = stacked
labels = false
color = lightgrey
[target gene span]
file = ${plot_dir}/target_gene.span.bed
title = target gene
height = 0.6
display = stacked
labels = true
fontsize = 8
color = darkblue
[other exons]
file = ${plot_dir}/other_genes.exon.bed
title = other exons
height = 1
display = stacked
labels = false
color = grey
[target gene exons]
file = ${plot_dir}/target_gene.exon.bed
title = target exons
height = 1.5
display = stacked
labels = false
color = darkblue
EOF
conda activate pygenometracks
pyGenomeTracks \
--tracks ${plot_dir}/${safe_id}.tracks.ini \
--region ${region} \
--outFileName ${plot_dir}/${safe_id}.pyGenomeTracks.${range}bp.pdf \
--width 40 \
--height 22 \
--dpi 300 \
--trackLabelFraction 0.22
bash plot_gene_hERV_chimeric_pgt.sh \
chr16:2663407:2671594 \
ENSG00000260565.10 \
HML4-16p13.3 \
internal/terminator \
15000
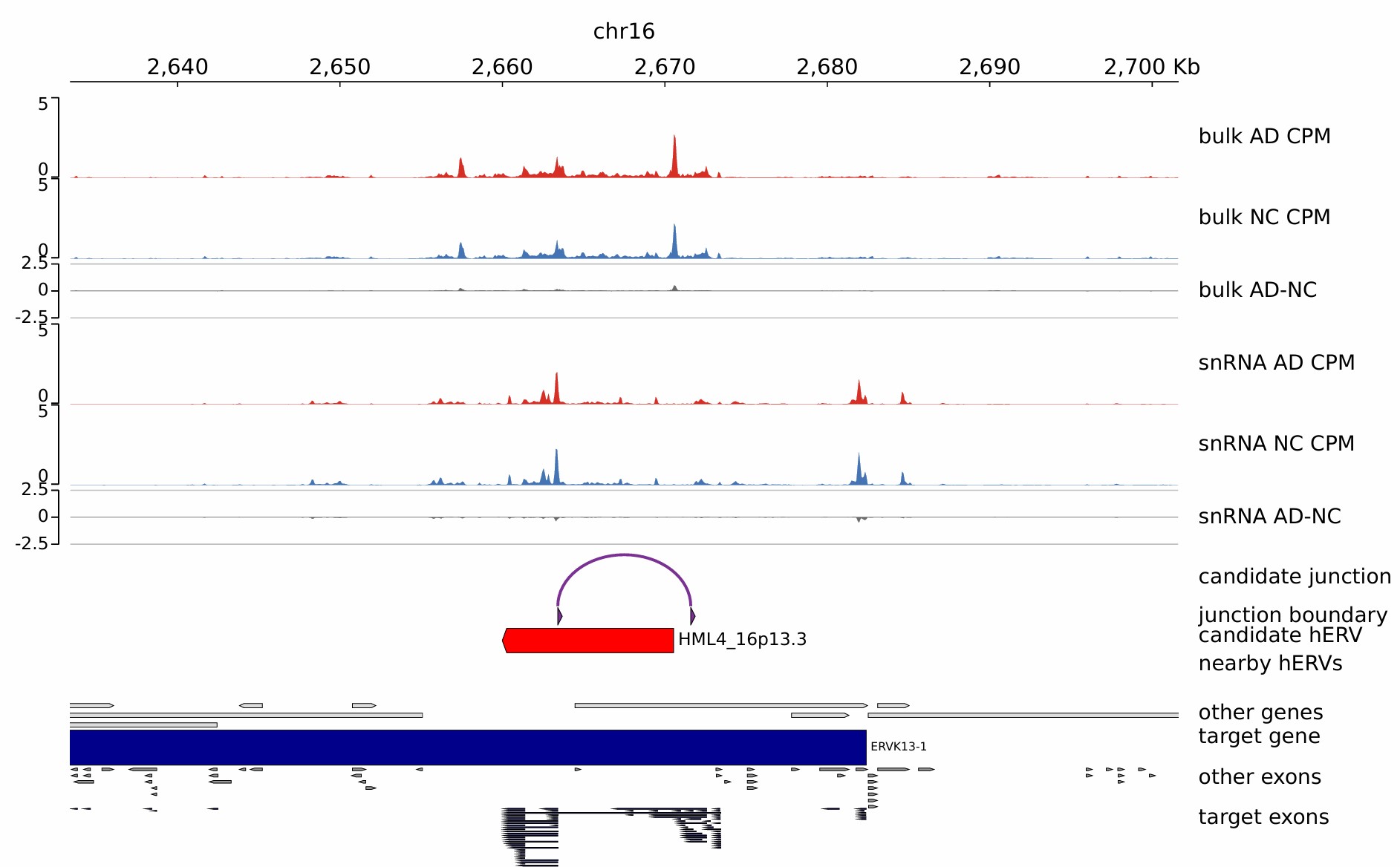
弧线是pipeline中生成的junction位置标记,与IGV里的RNA-seq junction弧线不同,它通常来自BAM中的剪接比对信息,表示某些reads真的跨过了一个剪接位点,而这里的弧线不包含read数量,也不表示这条junction在AD中有多少reads、NC中有多少reads
- 对于RNA-seq剪接junction来说,弧线通常连接两个被保留的read block,中间弧线跨过的区域往往是被剪掉的内含子区域
- 如果hERV位于弧线端点附近,可能支持hERV参与junction边界
- 如果hERV完全位于弧线中间,被弧线跨过去,则可能表示hERV所在区域被跳过
结合pyGenomeTracks画图分析后的候选位点:预览 | 下载
最核心的位点的pyGenomeTracks图:预览 | 下载 | 所有位点的图
这是我对全部候选运行画图函数后的结果,为了方便上传文件,我将每9张图以3*3的方式合成了一张大图,每张子图的上方标题标明其位点名。请仔细查看每一张子图,为我选出比较有意义的结果
我想把你上面给我的第一梯队和第二梯队整合到一个xlsx中(分成两个表),包括你上面表格里的内容,以及gene_hERV_chimeric_candidates.xlsx中这些位点的关键参数(选最关键的列,不要把全部列都合并上去)。我画图文件的文件名格式是chr21_20778547_20781348_ENSG00000224924.10_MER61-21q21.1b_MER61-21q21.1c_internal_terminator.pyGenomeTracks.15000bp这样,你可以先生成”你上面表格里的内容“的表格,然后给出与gene_hERV_chimeric_candidates.xlsx合并的R代码
other try
如果hERV和exon重叠,那么就是原先的转录本;嵌合转录本应该找hERV给基因提供了新exon(promoter 3’UTR等等):现在这套流程还不能证明这个hERV exon一定是新的,可能本来就在GENCODE/RefSeq里,那它可能只是正常转录本的一部分,不是新出现的isoform
- 也不能证明promoter_tss/promoter就是真正的alternative promoter(需要长读长数据)
- BigWig只能说明某个区域有reads覆盖,真正证明gene和hERV连接的是junction read
| 类型 | 结构解释 | 需要的核心证据 |
|---|---|---|
| hERV-derived promoter/5’ exon | hERV/LTR提供新的5’端外显子,接到下游gene exon |
promoter_tss/promoter、同链、AD中有junction read、NC少或无、最好有5’端证据 |
| hERV-derived internal exon | hERV作为中间外显子被纳入gene转录本 |
internal,最好同时有“上游gene exon→hERV”和“hERV→下游gene exon”两个junction |
| hERV-derived terminal exon/3’UTR | gene转录延伸到hERV,hERV作为最后外显子或3’UTR样区域 |
terminator、上游gene exon接到hERV、AD中更明显、最好有3’端/polyA或长读长证据 |
在这个pipeline的原论文A mouse-specific retrotransposon drives a conserved Cdk2ap1 isoform essential for development中,作者发现一个mouse-specific MT2B2 retrotransposon作为promoter,驱动了一个新的Cdk2ap1 isoform(Cdk2ap1DN)。这个isoform不是普通Cdk2ap1转录本,而是从MT2B2附近起始,然后剪接到Cdk2ap1 exon2,因此跳过了canonical exon1,产生N端截短的蛋白
- RNA-seq/junction发现:MT2B2-derived exon splice到Cdk2ap1 exon2
- 3’/5’RACE:确认TSS在MT2B2内部
- RT-PCR:确认MT2B2和Cdk2ap1 exon2之间存在剪接连接
- qPCR:比较canonical Cdk2ap1和MT2B2-driven Cdk2ap1DN的表达阶段
- CRISPR删除MT2B2:特异性消除Cdk2ap1DN表达,不影响邻近基因
- 功能实验:证明这个isoform影响胚胎发育和细胞增殖
原论文中过滤isoform是选preimplantation embryo中阶段特异的新isoform;而我这里应该是根据AD特异高或低来选?很多hERV-gene重叠可能本来就是正常脑组织转录本的一部分,也许需要看该junction是否已存在于GENCODE/RefSeq、hERV-derived exon是否是已知exon
-
比较canonical isoform:
hERV-derived exon → gene exon2-canonical exon1 → gene exon2如果AD中hERV→gene exon2上升,而canonical exon1→gene exon2不升甚至下降,就可能说明这个更可能是alternative promoter/isoform switching
//todo 嵌合转录本有没有新的方法(可以先用这套)
hERV与基因组结构变异
HERV的变化有可能是纯转录表观,也有可能跟基因组变化有关。这是一个AD队列里基因组结构变异跟HERV位点的overlap(来源于一个大队列的两个子队列分别算的结果),有没有跟转录信号能匹配起来的位点?
retrotransposon_SV_in_AD_cohort.xlsx:
- ID:SV事件ID,格式大致是
chr_start_length_type,例如chr19_19751989_47215_DUP -
chrom/start/end:SV所在染色体/起始坐标/终止坐标
注:虽然这个
length有的可能非常大,但这个确实是长度,因为这个就是表中start-end的值 - gene_id/gene_name:与SV重叠的HERV位点ID
- gene_start/gene_end:该HERV位点起始/终止坐标
-
AC_AD/AC_NC/AC_MCI/AC_AD_MCI:AD组/NC组/illumina中MCI组/huada中AD+MCI合并组中该SV的alternative allele count
alternative allele count:某个SV事件的非参考等位基因数量。参考基因组代表“没有这个变异”的状态,如果某个人带有这个SV,比如某段插入、缺失或重复,就记为alternative allele。所有样本的alternative allele数加起来就得到该值
个体基因型 含义 贡献的alternative allele count 0/0 不携带这个SV 0 0/1 杂合携带 1 1/1 纯合携带 2 MCI:mild cognitive impairment,轻度认知障碍。它常被看作正常衰老和AD之间的中间状态,但不是所有MCI都会发展成AD
AD+MCI合并组就是把AD样本和MCI样本放在一起作为一个疾病相关组,再和NC比较。这样做的目的通常是提高样本量和检验能力,但代价是表型更混合
比较方式 含义 特点 ADvsNC AD和正常对照比较 表型更明确 MCIvsNC MCI和正常对照比较 看早期/中间状态 AD+MCIvsNC AD和MCI合并后与正常对照比较 样本量更大,但疾病状态更混合 - xxx_sample_counts:样本数
-
xx_vs_yy_OR:xx-yy odds ratio
ADvsNC odds ratio:这个SV在AD组中出现的优势,和在NC组中出现的优势相比,有多大
AD组100人,AC_AD=20 NC组100人,AC_NC=10 ↓ AD组reference allele = 200 - 20 = 180 NC组reference allele = 200 - 10 = 190 ↓ OR = (20/180) / (10/190) = 2.11OR值 含义 OR>1 这个SV在AD组中更常见,可能是AD富集 OR<1 这个SV在AD组中更少见,可能在NC中更常见 OR=1 AD和NC中频率差不多 注:OR=2不等于AD组携带人数是NC组2倍。如果SV很稀有,OR和频率比会比较接近;如果SV不稀有,二者会有明显差别
某个HERV位点是否同时满足“基因组SV层面有重叠”和“转录层面有AD差异表达”
- 用
DE.xlsx中的all_MAST筛显著转录信号,p_val_adj<0.05作为显著 - 将SV-HERV overlap表与DE显著HERV取交集
-
计算等位基因频率(AF_AD/AF_NC)、log2OR_AD_vs_NC_corr、Fisher检验P值以及BH校正后q值
AD组100人,AC_AD=20 NC组50人,AC_NC=20 虽然两个组AC都是20,但频率不同: AF_AD = 20 / 200 = 0.10 AF_NC = 20 / 100 = 0.20 说明这个SV其实在NC中更常见,而不是AD和NC一样多log2OR_AD_vs_NC_corr:这个SV是AD富集,还是AD减少?方向和幅度大概是多少?
log2OR_AD_vs_NC_corr大致含义 2 SV在AD中的优势约为NC的4倍 1 SV在AD中的优势约为NC的2倍 0 AD和NC差不多 -1 SV在AD中的优势约为NC的一半 -2 SV在AD中的优势约为NC的四分之一 Fisher检验:AD组和NC组中这个SV的等位基因比例是否显著不同
q值:在这些显著结果中,预计有多大比例可能是假阳性
- 粗略判断SV方向和表达方向是否一致:
-
DUP/INS在AD富集,则预期HERV表达升高。DUP是duplication重复,INS是insertion插入,如果一个HERV位点附近或内部发生DUP/INS:- HERV拷贝数增加
- HERV附近调控序列增加
- 插入带来新的启动子、增强子或剪接结构
- 这个区域更容易产生HERV相关转录
-
DEL在AD富集,则预期HERV表达降低。DEL是deletion缺失,如果一个缺失覆盖了HERV位点,或者覆盖了HERV启动子/调控区域,那么这个HERV转录可能减少 - SV对HERV表达的影响不一定这么简单,所以只能粗略看一下:
情况 可能导致的问题 SV只覆盖HERV的一小段 不一定影响转录 SV覆盖的是HERV内部而不是启动区域 方向可能不清楚 DEL删除的是抑制元件 反而可能让表达升高 DUP破坏局部染色质结构 不一定增加表达 SV很稀有 频率差异可能不稳定 表达是细胞类型特异的 bulk层面的SV频率不一定能解释某个细胞类型的HERV变化 SV表是群体汇总数据 不能知道“携带SV的样本”是否真的表达改变 -
import re
import numpy as np
import pandas as pd
from scipy.stats import fisher_exact
def bh_adjust(p):
p = pd.Series(p, dtype="float64")
q = pd.Series(np.nan, index=p.index, dtype="float64")
ok = p.notna()
if ok.sum() == 0:
return q
x = p[ok].values
order = np.argsort(x)
ranked = x[order] * len(x) / np.arange(1, len(x) + 1)
ranked = np.minimum.accumulate(ranked[::-1])[::-1]
q.loc[p[ok].index[order]] = np.minimum(ranked, 1)
return q
def sv_type(x):
m = re.search(r"_(INS|DEL|DUP|INV|BND)$", str(x))
return m.group(1) if m else np.nan
def de_desc(g, n=5):
g = g.sort_values("p_val_adj").head(n)
return "; ".join([f"{r.DE_sheet}/{r.celltype}:{r.avg_log2FC:.2f},padj={r.p_val_adj:.1e}" for r in g.itertuples()])
def all_mast_desc(g):
g = g[g["DE_sheet"].eq("all_MAST")].sort_values("p_val_adj")
if len(g) == 0:
return np.nan
return "; ".join([f"{r.celltype}:{r.avg_log2FC:.2f},padj={r.p_val_adj:.1e}" for r in g.itertuples()])
# 读取SV-HERV overlap文件
sv_all = []
for cohort, sheet in [("huada", "huada"), ("illumina", "illumina")]:
x = pd.read_excel(r"C:\Users\17185\Desktop\Paper_reading_ChenLab\retrotransposon_SV_in_AD_cohort.xlsx", sheet_name=sheet)
if cohort == "huada":
x = x.rename(columns={
"gene_id": "feature_raw",
"AC_AD_MCI": "AC_MCI",
"AD_sample_counts": "n_AD",
"NC_sample_counts": "n_NC",
"AD_MCI_sample_counts": "n_MCI"
})
else:
x = x.rename(columns={
"gene_name": "feature_raw",
"AD_sample_counts": "n_AD",
"NC_sample_counts": "n_NC",
"MCI_sample_counts": "n_MCI"
})
x["cohort"] = cohort
x["feature"] = x["feature_raw"].astype(str).str.replace("_", "-", regex=False)
x["sv_type"] = x["ID"].map(sv_type)
x["sv_len"] = x["end"] - x["start"]
x["sv_herv_overlap_bp"] = (
np.minimum(x["end"], x["gene_end"]) - np.maximum(x["start"], x["gene_start"])
).clip(lower=0)
x["sv_herv_overlap_frac"] = x["sv_herv_overlap_bp"] / (x["gene_end"] - x["gene_start"])
x["AF_AD"] = x["AC_AD"] / (2 * x["n_AD"])
x["AF_NC"] = x["AC_NC"] / (2 * x["n_NC"])
x["AF_MCI"] = x["AC_MCI"] / (2 * x["n_MCI"])
x["log2OR_AD_vs_NC_corr"] = np.log2(
((x["AC_AD"] + 0.5) / (2*x["n_AD"] - x["AC_AD"] + 0.5)) /
((x["AC_NC"] + 0.5) / (2*x["n_NC"] - x["AC_NC"] + 0.5))
)
x["log2OR_MCI_vs_NC_corr"] = np.log2(
((x["AC_MCI"] + 0.5) / (2*x["n_MCI"] - x["AC_MCI"] + 0.5)) /
((x["AC_NC"] + 0.5) / (2*x["n_NC"] - x["AC_NC"] + 0.5))
)
keep = [
"cohort", "feature", "ID", "sv_type", "chrom", "start", "end", "sv_len",
"gene_start", "gene_end", "sv_herv_overlap_bp", "sv_herv_overlap_frac",
"AC_AD", "AC_NC", "AC_MCI", "n_AD", "n_NC", "n_MCI",
"AF_AD", "AF_NC", "AF_MCI",
"log2OR_AD_vs_NC_corr", "log2OR_MCI_vs_NC_corr"
]
sv_all.append(x[keep])
sv = pd.concat(sv_all, ignore_index=True)
# 读取DE文件
de_list = []
for sheet in pd.ExcelFile(r"C:\Users\17185\Desktop\file\ATACtest\res\DE.xlsx").sheet_names:
x = pd.read_excel(r"C:\Users\17185\Desktop\file\ATACtest\res\DE.xlsx", sheet_name=sheet)
x["DE_sheet"] = sheet
de_list.append(x)
de = pd.concat(de_list, ignore_index=True)
sig_de = de[de["p_val_adj"] < 0.05].copy()
best_de = sig_de.sort_values("p_val_adj").drop_duplicates("feature")
best_de = best_de[
["feature", "DE_sheet", "celltype", "avg_log2FC", "p_val_adj"]
].rename(columns={
"DE_sheet": "best_DE_sheet",
"celltype": "best_DE_celltype",
"avg_log2FC": "best_DE_log2FC",
"p_val_adj": "best_DE_p_adj"
})
de_summary = sig_de.groupby("feature").agg({
"DE_sheet": lambda z: ",".join(sorted(z.dropna().astype(str).unique())),
"celltype": lambda z: ",".join(sorted(z.dropna().astype(str).unique()))
}).reset_index()
de_summary = de_summary.rename(columns={
"DE_sheet": "sig_sheets",
"celltype": "sig_celltypes"
})
n_sig = sig_de.groupby("feature").size().reset_index(name="n_sig_de_tests")
de_summary = de_summary.merge(n_sig, on="feature", how="left")
de_summary = de_summary.merge(best_de, on="feature", how="left")
all_mast_features = set(sig_de.loc[sig_de["DE_sheet"].eq("all_MAST"), "feature"])
de_summary["is_all_MAST_sig"] = de_summary["feature"].isin(all_mast_features)
de_summary = de_summary.merge(
sig_de.groupby("feature").apply(all_mast_desc).rename("all_MAST_desc").reset_index(),
on="feature", how="left"
)
de_summary = de_summary.merge(
sig_de.groupby("feature").apply(de_desc).rename("top_DE_desc").reset_index(),
on="feature", how="left"
)
# 两者匹配
matched = sv.merge(de_summary, on="feature", how="inner")
matched["fisher_p_AD_vs_NC"] = [
fisher_exact([[a, 2*na-a], [c, 2*nc-c]])[1]
for a, na, c, nc in zip(matched["AC_AD"], matched["n_AD"], matched["AC_NC"], matched["n_NC"])
]
matched["fisher_q_AD_vs_NC_in_matched"] = bh_adjust(matched["fisher_p_AD_vs_NC"])
expected = np.sign(matched["log2OR_AD_vs_NC_corr"])
expected = expected.where(~matched["sv_type"].eq("DEL"), -expected)
expected = expected.where(matched["sv_type"].isin(["DUP", "INS", "DEL"]), np.nan)
matched["simplified_expected_expr_sign_from_SV"] = expected
matched["direction_match_simplified"] = np.sign(matched["best_DE_log2FC"]).eq(
matched["simplified_expected_expr_sign_from_SV"]
)
matched = matched.sort_values([
"fisher_q_AD_vs_NC_in_matched", "best_DE_p_adj", "feature"
])
# 汇总结果
def sv_type_text(z):
return ";".join([f"{k}:{v}" for k, v in z.value_counts(sort=False).items()])
def one_feature_one_cohort(g):
top = g.loc[g["log2OR_AD_vs_NC_corr"].abs().idxmax()]
return pd.Series({
"n_sv": len(g),
"n_recurrent_sv_ACge5": int(((g["AC_AD"] + g["AC_NC"] + g["AC_MCI"]) >= 5).sum()),
"n_AD_enriched_sv": int((g["log2OR_AD_vs_NC_corr"] > 0).sum()),
"n_AD_depleted_sv": int((g["log2OR_AD_vs_NC_corr"] < 0).sum()),
"max_abs_log2OR_ADvsNC": g["log2OR_AD_vs_NC_corr"].abs().max(),
"top_SV": top["ID"],
"top_sv_type": top["sv_type"],
"top_log2OR_ADvsNC": top["log2OR_AD_vs_NC_corr"],
"top_AF_AD": top["AF_AD"],
"top_AF_NC": top["AF_NC"],
"top_AC_AD": top["AC_AD"],
"top_AC_NC": top["AC_NC"],
"top_fisher_p": top["fisher_p_AD_vs_NC"],
"sv_types": sv_type_text(g["sv_type"])
})
sv_sum = matched.groupby(["feature", "cohort"]).apply(one_feature_one_cohort).reset_index()
sv_wide = None
for cohort in ["huada", "illumina"]:
tmp = sv_sum[sv_sum["cohort"].eq(cohort)].drop(columns="cohort")
tmp = tmp.rename(columns={c: f"{cohort}_{c}" for c in tmp.columns if c != "feature"})
sv_wide = tmp if sv_wide is None else sv_wide.merge(tmp, on="feature", how="outer")
cohorts = matched.groupby("feature")["cohort"].apply(
lambda z: ",".join(sorted(z.unique()))
).reset_index(name="SV_cohorts")
cohorts["SV_in_both_cohorts"] = (
cohorts["SV_cohorts"].str.contains("huada") &
cohorts["SV_cohorts"].str.contains("illumina")
)
same_n = matched.groupby(["feature", "ID"])["cohort"].nunique().reset_index(name="n_cohorts")
same_n = same_n[same_n["n_cohorts"].eq(2)]
same_n = same_n.groupby("feature").size().reset_index(name="same_variant_in_both_cohorts_n")
# 合并HERV注释和gene相关性信息
overlap = pd.read_excel(r"C:\Users\17185\Desktop\file\ATACtest\res\hERV_overlap_expression.xlsx", sheet="hERV_overlap_expression_all_cel")
overlap = overlap[
["herv_id_expr", "chr", "start", "end", "overlap_type"]
].rename(columns={
"herv_id_expr": "feature",
"start": "herv_start",
"end": "herv_end"
})
corr = pd.read_excel(r"C:\Users\17185\Desktop\file\ATACtest\res\hERV_related_gene_expression_corr.xlsx")
corr = corr[
["herv_id_expr", "gene_name", "gene_type", "gene_choice", "loc_class",
"strand_relation", "overlap_relation", "nearest_gene_distance", "rho", "p_adj"]
].rename(columns={
"herv_id_expr": "feature",
"gene_name": "corr_gene_name",
"gene_type": "corr_gene_type",
"gene_choice": "corr_gene_choice",
"loc_class": "corr_loc_class",
"strand_relation": "corr_strand_relation",
"overlap_relation": "corr_overlap_relation",
"nearest_gene_distance": "corr_nearest_gene_distance",
"rho": "corr_rho",
"p_adj": "corr_p_adj"
})
# 总表
candidate_all = de_summary.merge(cohorts, on="feature", how="inner")
candidate_all = candidate_all.merge(sv_wide, on="feature", how="left")
candidate_all = candidate_all.merge(same_n, on="feature", how="left")
candidate_all["same_variant_in_both_cohorts_n"] = candidate_all["same_variant_in_both_cohorts_n"].fillna(0).astype(int)
candidate_all = candidate_all.merge(overlap, on="feature", how="left")
candidate_all = candidate_all.merge(corr, on="feature", how="left")
def get_priority(r):
if r["SV_in_both_cohorts"] and r["is_all_MAST_sig"]:
return "A_both_SV_and_all_MAST_DE"
if r["SV_in_both_cohorts"]:
return "B_both_SV_and_any_DE"
if r["is_all_MAST_sig"]:
return "C_one_SV_and_all_MAST_DE"
return "D_one_SV_and_any_DE"
candidate_all["priority"] = candidate_all.apply(get_priority, axis=1)
priority_order = {
"A_both_SV_and_all_MAST_DE": 1,
"B_both_SV_and_any_DE": 2,
"C_one_SV_and_all_MAST_DE": 3,
"D_one_SV_and_any_DE": 4
}
candidate_all["priority_rank"] = candidate_all["priority"].map(priority_order)
candidate_all = candidate_all.sort_values(
["priority_rank", "best_DE_p_adj", "feature"]
).drop(columns="priority_rank")
# 筛选表:两个SV子队列都有SV重叠 + all_MAST显著
candidate_filtered = candidate_all[
candidate_all["priority"].eq("A_both_SV_and_all_MAST_DE")
].copy()
with pd.ExcelWriter(r"C:\Users\17185\Desktop\file\ATACtest\res\HERV_SV_transcription_match_simple.xlsx") as writer:
candidate_all.to_excel(writer, sheet_name="candidate_all", index=False)
candidate_filtered.to_excel(writer, sheet_name="candidate_filtered", index=False)
| 指标 | 数量 |
|---|---|
| 两个SV子队列共享的HERV位点数 | 1703 |
| 任一DE表中显著的HERV位点数 | 298 |
| all_MAST显著的HERV位点数 | 69 |
| SV重叠且任一DE显著的HERV位点数 | 225 |
| 两个SV子队列均有重叠且all_MAST显著的HERV位点数 | 9 |
| 同一个SV ID+HERV在两个子队列都出现的记录数 | 19 |
| 匹配结果中Fisher p<0.05的SV-HERV记录数 | 67 |
| 匹配结果中BH校正q<0.05的SV-HERV记录数 | 3 |
298个显著差异HERV中有225个能找到SV重叠,但考虑差异显著后,只有少数HERV转录变化可能与SV相关:
- 预览 | 下载
-
三个q显著结果其实是同一个SV区域:一个覆盖17q21.31-17q21.32附近多个HERV的DUP在AD中频率低于NC,并且对应HERV在转录层面表现为AD下调
注:这种超大SV在后续分析中要谨慎:一个39.8Mb的DUP会覆盖非常多基因、HERV和调控区域。它和某个HERV overlap,不一定说明它专门影响这个HERV;更可能是一个大段CNV把很多位点都一起覆盖了
- 9个两个SV子队列均有重叠且all_MAST显著的HERV位点
差异表达的HERV位点的reads覆盖情况
产出的是短eRNA(比如5’LTR上)还是长lncRNA?别人bulkRNA结果中与我scRNA结果中方向相反的差异表达hERV位点,产出的是短的还是长的
- eRNA:增强子RNA,即增强子转录的ncRNA,长度为0.5-5kb,因此可以分类为lncRNA
- 短eRNA-like信号:reads主要集中在HERV的5’LTR或soloLTR附近,覆盖范围很短,没有明显向HERV内部、3’端或邻近基因延伸。说明这个LTR可能像增强子/启动子附近产生的短RNA一样,只产生局部转录信号
- 长lncRNA/长转录本信号:reads不是只堆在5’LTR,而是从LTR区域继续延伸到HERV内部、3’LTR、下游基因间区,或者进入邻近基因/lncRNA外显子。说明它可能不是一个局部短转录,而是一个较长的HERV相关转录本
1.准备候选位点和barcode文件:
library(Seurat)
library(tidyverse)
library(readxl)
library(data.table)
res_dir <- "/public/home/GENE_proc/wth/GSE174367/0530test/hERV_coverage/"
load("/public/home/GENE_proc/wth/benkebishe/data/all_final.RData"); seu=all; rm(all)
meta <- seu@meta.data %>% rownames_to_column("cell")
meta <- meta %>%
mutate(
GSM = as.character(GSM),
dataset = as.character(dataset),
group = as.character(group),
celltype = as.character(celltype),
barcode = str_extract(cell, "[ACGT]{16}(-[0-9]+)?$"),
barcode = str_remove(barcode, "-[0-9]+$")
) %>%
filter(group %in% c("AD", "NC"), !is.na(barcode), !is.na(GSM), !is.na(dataset), !is.na(celltype))
de <- read_xlsx("/public/home/GENE_proc/wth/GSE174367/0530test/res/DE.xlsx", sheet = "all_MAST") %>% as.data.frame()
de2 <- de %>%
mutate(
locus_raw = feature,
locus_id = str_replace_all(feature, "-", "_"),
AD_pct = pct.1,
NC_pct = pct.2,
expressed_in_ct = AD_pct > 0 | NC_pct > 0,
direction = if_else(avg_log2FC > 0, "AD_up", "AD_down"),
assumed_bulk_sc_opposite = avg_log2FC < 0
) %>%
filter(p_val_adj < 0.05, expressed_in_ct) %>%
arrange(celltype, p_val_adj) %>%
group_by(celltype) %>%
ungroup()
bed <- fread("/public/home/GENE_proc/wth/GSE174367/0530test/data/hERV_locus.bed", col.names = c("chr", "start", "end", "locus_id", "score", "strand"))
candidate <- de2 %>%
left_join(bed, by = "locus_id") %>%
mutate(celltype_safe = str_replace_all(celltype, "[^A-Za-z0-9_.-]+", "_"))
candidate <- candidate %>%
filter(!is.na(chr), chr %in% paste0("chr", c(1:22, "X", "Y"))) %>%
mutate(
start = as.integer(start),
end = as.integer(end),
win_start = pmax(start - 50000, 0),
win_end = end + 50000
)
write.csv(candidate, file.path(res_dir, "candidate_hERV_DE_with_coord.csv"), row.names = FALSE)
candidate_loci <- candidate %>%
distinct(locus_id, chr, start, end, strand)
candidate_window_bed <- candidate_loci %>%
mutate(win_start = pmax(start - 50000, 0), win_end = end + 50000) %>%
transmute(chr, win_start, win_end, locus_id, score = 0, strand)
write.table(candidate_window_bed, file.path(res_dir, "candidate_hERV_window50k.bed"),
sep = "\t", quote = FALSE, row.names = FALSE, col.names = FALSE)
five_len <- 700
part_5p <- candidate_loci %>%
mutate(
part_start = if_else(strand == "+", start, pmax(end - five_len, start)),
part_end = if_else(strand == "+", pmin(start + five_len, end), end),
region_type = "approx_5p_700bp"
)
part_body <- candidate_loci %>%
mutate(
part_start = if_else(strand == "+", pmin(start + five_len, end), start),
part_end = if_else(strand == "+", end, pmax(end - five_len, start)),
region_type = "body_rest"
)
part_up5 <- candidate_loci %>%
mutate(
part_start = if_else(strand == "+", pmax(start - 10000, 0), end),
part_end = if_else(strand == "+", start, end + 10000),
region_type = "upstream_5p_10kb"
)
part_down3 <- candidate_loci %>%
mutate(
part_start = if_else(strand == "+", end, pmax(start - 10000, 0)),
part_end = if_else(strand == "+", end + 10000, start),
region_type = "downstream_3p_10kb"
)
parts <- bind_rows(part_5p, part_body, part_up5, part_down3) %>%
filter(part_end > part_start) %>%
transmute(chr, part_start, part_end, name = paste(locus_id, region_type, sep = "|"), score = 0, strand)
write.table(parts, file.path(res_dir, "candidate_hERV_parts_approx5p.bed"),
sep = "\t", quote = FALSE, row.names = FALSE, col.names = FALSE)
target_ct <- unique(candidate$celltype)
meta2 <- meta %>%
filter(celltype %in% target_ct) %>%
mutate(celltype_safe = str_replace_all(celltype, "[^A-Za-z0-9_.-]+", "_"))
bc_dir <- file.path(res_dir, "barcodes")
dir.create(bc_dir, recursive = TRUE, showWarnings = FALSE)
barcode_index <- meta2 %>%
group_by(dataset, GSM, group, celltype, celltype_safe) %>%
summarise(n_cell = n_distinct(cell), barcodes = list(sort(unique(barcode))), .groups = "drop") %>%
filter(n_cell > 0)
barcode_index$barcode_file <- NA_character_
for(i in seq_len(nrow(barcode_index))){
out_dir <- file.path(bc_dir, barcode_index$dataset[i], barcode_index$GSM[i])
dir.create(out_dir, recursive = TRUE, showWarnings = FALSE)
out_file <- file.path(out_dir, paste(barcode_index$celltype_safe[i], barcode_index$group[i], "barcodes.txt", sep = "."))
writeLines(barcode_index$barcodes[[i]], out_file)
barcode_index$barcode_file[i] <- out_file
}
barcode_index_out <- barcode_index %>% select(dataset, GSM, group, celltype, celltype_safe, n_cell, barcode_file)
fwrite(barcode_index_out, file.path(res_dir, "barcode_file_map.tsv"), sep = "\t")
2.按celltype/group生成pseudobulk BAM和BigWig
module load miniconda3/base
conda create -n hERV_coverage deeptools samtools bedtools -y
conda activate hERV_coverage
stellarscope的bam:
# cd /public/home/GENE_proc/wth/GSE174367/stellarscope/
# find . -mindepth 2 -maxdepth 2 ! -name 'stellarscope-updated.bam' -exec rm -r -- {} +
# mv * /public/home/GENE_proc/wth/GSE174367/0530test/stellarscope_res/
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/hERV_coverage/
ss_dir=/public/home/GENE_proc/wth/GSE174367/0530test/stellarscope_res/
map_tsv=${res_dir}/barcode_file_map.tsv
window_bed=${res_dir}/candidate_hERV_window50k.bed
parts_bed=${res_dir}/candidate_hERV_parts_approx5p.bed
threads=16
mode=ss_avg
tmp_dir=${res_dir}/tmp_bam_${mode}
pseudo_dir=${res_dir}/pseudobam_${mode}
bw_dir=${res_dir}/bigwig_${mode}
cov_dir=${res_dir}/coverage_parts_${mode}
mkdir -p ${tmp_dir} ${pseudo_dir} ${bw_dir} ${cov_dir}
sample_stat=${res_dir}/sample_bam_read_counts_${mode}.tsv
echo -e "dataset\tGSM\tgroup\tcelltype\tcelltype_safe\tn_cell\ttotal_assigned_alignments\tregion_bam" > ${sample_stat}
tail -n +2 ${map_tsv} | while IFS=$'\t' read -r dataset GSM group celltype celltype_safe n_cell barcode_file; do
bam=${ss_dir}/${GSM}/stellarscope-updated.bam
if [[ ! -f "${bam}" ]]; then
echo "[WARN] Missing BAM: ${bam}" >&2
continue
fi
out_bam=${tmp_dir}/${dataset}.${GSM}.${celltype_safe}.${group}.${mode}.bam
echo "[INFO] ${dataset} ${GSM} ${celltype_safe} ${group}"
total=$(samtools view -@ ${threads} -c -F 4 -D CB:${barcode_file} ${bam})
if [[ "${total}" == "0" ]]; then
echo "[WARN] No assigned alignments: ${dataset} ${GSM} ${celltype_safe} ${group}" >&2
continue
fi
samtools view -@ ${threads} -h -F 4 -D CB:${barcode_file} ${bam} \
| samtools view -@ ${threads} -b -L ${window_bed} - \
| samtools sort -@ ${threads} -o ${out_bam} -
region_count=$(samtools view -@ ${threads} -c ${out_bam})
if [[ "${region_count}" == "0" ]]; then
rm -f ${out_bam}
echo "[WARN] No reads in candidate windows: ${dataset} ${GSM} ${celltype_safe} ${group}" >&2
continue
fi
samtools index -@ ${threads} ${out_bam}
echo -e "${dataset}\t${GSM}\t${group}\t${celltype}\t${celltype_safe}\t${n_cell}\t${total}\t${out_bam}" >> ${sample_stat}
done
pseudo_stat=${res_dir}/pseudobam_read_counts_${mode}.tsv
echo -e "pseudo_id\tcelltype_safe\tgroup\tmode\ttotal_assigned_alignments\tbam\tbigwig" > ${pseudo_stat}
tail -n +2 ${sample_stat} | cut -f3,5 | sort -u | while IFS=$'\t' read -r group celltype_safe; do
mapfile -t files < <(awk -F'\t' -v g="${group}" -v ct="${celltype_safe}" '$3==g && $5==ct {print $8}' ${sample_stat})
total=$(awk -F'\t' -v g="${group}" -v ct="${celltype_safe}" '$3==g && $5==ct {s+=$7} END{print s+0}' ${sample_stat})
if [[ ${#files[@]} -eq 0 || "${total}" == "0" ]]; then
continue
fi
pseudo_id=all.${celltype_safe}.${group}.${mode}
out_bam=${pseudo_dir}/${pseudo_id}.bam
out_bw=${bw_dir}/${pseudo_id}.bw
if [[ ${#files[@]} -eq 1 ]]; then
cp "${files[0]}" ${out_bam}
else
samtools merge -@ ${threads} -f ${out_bam} "${files[@]}"
fi
samtools index -@ ${threads} ${out_bam}
scale=$(awk -v n=${total} 'BEGIN{printf "%.10f", 1000000/n}')
bamCoverage --bam ${out_bam} -o ${out_bw} -of bigwig --binSize 10 --normalizeUsing None --scaleFactor ${scale} -p ${threads}
bedtools coverage -counts -a ${parts_bed} -b ${out_bam} > ${cov_dir}/${pseudo_id}.parts.tsv
echo -e "${pseudo_id}\t${celltype_safe}\t${group}\t${mode}\t${total}\t${out_bam}\t${out_bw}" >> ${pseudo_stat}
done
STAR的bam:
res_dir=/public/home/GENE_proc/wth/GSE174367/0530test/hERV_coverage/
bam_dir_157827=/public/home/GENE_proc/wth/GSE157827/star_bam
bam_dir_174367=/public/home/GENE_proc/wth/GSE174367/star_bam
map_tsv=${res_dir}/barcode_file_map.tsv
window_bed=${res_dir}/candidate_hERV_window50k.bed
threads=16
mode=star
tmp_dir=${res_dir}/tmp_bam_${mode}
pseudo_dir=${res_dir}/pseudobam_${mode}
bw_dir=${res_dir}/bigwig_${mode}
mkdir -p ${tmp_dir} ${pseudo_dir} ${bw_dir}
sample_stat=${res_dir}/sample_bam_read_counts_${mode}.tsv
echo -e "dataset\tGSM\tgroup\tcelltype\tcelltype_safe\tn_cell\ttotal_alignments\tregion_bam" > ${sample_stat}
tail -n +2 ${map_tsv} | while IFS=$'\t' read -r dataset GSM group celltype celltype_safe n_cell barcode_file; do
if [[ "${dataset}" == "GSE157827" ]]; then
bam_dir=${bam_dir_157827}
else
bam_dir=${bam_dir_174367}
fi
bam=${bam_dir}/${GSM}Aligned.sortedByCoord.out.bam
if [[ ! -f "${bam}" ]]; then
echo "[WARN] Missing BAM: ${bam}" >&2
continue
fi
if [[ ! -f "${bam}.bai" ]]; then
samtools index -@ ${threads} ${bam}
fi
out_bam=${tmp_dir}/${dataset}.${GSM}.${celltype_safe}.${group}.${mode}.bam
echo "[INFO] ${dataset} ${GSM} ${celltype_safe} ${group}"
total=$(samtools view -@ ${threads} -c -F 260 -D CB:${barcode_file} ${bam})
if [[ "${total}" == "0" ]]; then
echo "[WARN] No STAR alignments: ${dataset} ${GSM} ${celltype_safe} ${group}" >&2
continue
fi
samtools view -@ ${threads} -b -F 260 -D CB:${barcode_file} -L ${window_bed} ${bam} \
| samtools sort -@ ${threads} -o ${out_bam} -
region_count=$(samtools view -@ ${threads} -c ${out_bam})
if [[ "${region_count}" == "0" ]]; then
rm -f ${out_bam}
echo "[WARN] No reads in candidate windows: ${dataset} ${GSM} ${celltype_safe} ${group}" >&2
continue
fi
samtools index -@ ${threads} ${out_bam}
echo -e "${dataset}\t${GSM}\t${group}\t${celltype}\t${celltype_safe}\t${n_cell}\t${total}\t${out_bam}" >> ${sample_stat}
done
pseudo_stat=${res_dir}/pseudobam_read_counts_${mode}.tsv
echo -e "pseudo_id\tcelltype_safe\tgroup\tmode\ttotal_alignments\tbam\tbigwig" > ${pseudo_stat}
tail -n +2 ${sample_stat} | cut -f3,5 | sort -u | while IFS=$'\t' read -r group celltype_safe; do
mapfile -t files < <(awk -F'\t' -v g="${group}" -v ct="${celltype_safe}" '$3==g && $5==ct {print $8}' ${sample_stat})
total=$(awk -F'\t' -v g="${group}" -v ct="${celltype_safe}" '$3==g && $5==ct {s+=$7} END{print s+0}' ${sample_stat})
if [[ ${#files[@]} -eq 0 || "${total}" == "0" ]]; then
continue
fi
pseudo_id=all.${celltype_safe}.${group}.${mode}
out_bam=${pseudo_dir}/${pseudo_id}.bam
out_bw=${bw_dir}/${pseudo_id}.bw
if [[ ${#files[@]} -eq 1 ]]; then
cp "${files[0]}" ${out_bam}
else
samtools merge -@ ${threads} -f ${out_bam} "${files[@]}"
fi
samtools index -@ ${threads} ${out_bam}
scale=$(awk -v n=${total} 'BEGIN{printf "%.10f", 1000000/n}')
bamCoverage --bam ${out_bam} -o ${out_bw} -of bigwig --binSize 10 --normalizeUsing None --scaleFactor ${scale} -p ${threads}
echo -e "${pseudo_id}\t${celltype_safe}\t${group}\t${mode}\t${total}\t${out_bam}\t${out_bw}" >> ${pseudo_stat}
done
3. 汇总覆盖特征并粗分类
library(data.table)
library(dplyr)
library(tidyr)
library(stringr)
library(readr)
res_dir <- "/public/home/GENE_proc/wth/GSE174367/0530test/hERV_coverage/"
mode <- "ss_avg"
candidate_file <- file.path(res_dir, "candidate_hERV_DE_with_coord.csv")
pseudo_stat_file <- file.path(res_dir, paste0("pseudobam_read_counts_", mode, ".tsv"))
cov_dir <- file.path(res_dir, paste0("coverage_parts_", mode))
candidate <- read.csv(candidate_file) %>%
mutate(celltype_safe = str_replace_all(celltype, "[^A-Za-z0-9_.-]+", "_"))
pseudo_stat <- fread(pseudo_stat_file) %>% as.data.frame()
if("total_assigned_alignments" %in% colnames(pseudo_stat)){
pseudo_stat <- pseudo_stat %>%
rename(total_mapped = total_assigned_alignments)
}
cov_files <- list.files(cov_dir, pattern = "\\.parts\\.tsv$", full.names = TRUE)
cov_list <- lapply(cov_files, function(f){
x <- fread(f, header = FALSE)
setnames(x, c("chr", "start", "end", "part_name", "score", "strand", "read_count"))
x$pseudo_id <- str_remove(basename(f), "\\.parts\\.tsv$")
x
})
cov <- bind_rows(cov_list) %>%
mutate(
width = end - start,
locus_id = str_split_fixed(part_name, "\\|", 2)[, 1],
region_type = str_split_fixed(part_name, "\\|", 2)[, 2]
) %>%
left_join(pseudo_stat, by = "pseudo_id") %>%
mutate(
cpm = read_count / total_mapped * 1e6,
cpm_per_kb = cpm / (width / 1000)
)
wide_density <- cov %>%
select(pseudo_id, celltype_safe, group, mode, locus_id, region_type, cpm_per_kb) %>%
pivot_wider(names_from = region_type, values_from = cpm_per_kb, values_fill = 0)
wide_count <- cov %>%
select(pseudo_id, celltype_safe, group, mode, locus_id, region_type, read_count) %>%
pivot_wider(names_from = region_type, values_from = read_count, values_fill = 0, names_prefix = "raw_")
summary <- wide_density %>%
left_join(wide_count, by = c("pseudo_id", "celltype_safe", "group", "mode", "locus_id")) %>%
mutate(
total_herv_raw = raw_approx_5p_700bp + raw_body_rest,
total_region_raw = raw_approx_5p_700bp + raw_body_rest + raw_upstream_5p_10kb + raw_downstream_3p_10kb,
fivep_to_body_density_ratio = approx_5p_700bp / (body_rest + 1e-6),
downstream_to_herv_density_ratio = downstream_3p_10kb / (pmax(approx_5p_700bp, body_rest) + 1e-6),
upstream_to_herv_density_ratio = upstream_5p_10kb / (pmax(approx_5p_700bp, body_rest) + 1e-6),
coverage_class = case_when(
total_region_raw < 10 ~ "reads_too_few",
approx_5p_700bp > 0 & fivep_to_body_density_ratio >= 2 & downstream_to_herv_density_ratio < 0.3 ~ "5p_limited_short_like",
body_rest > 0 & body_rest >= 0.5 * approx_5p_700bp & downstream_to_herv_density_ratio < 0.3 ~ "HERV_body_covered",
downstream_to_herv_density_ratio >= 0.3 ~ "extended_to_3p_flank",
upstream_to_herv_density_ratio >= 0.5 ~ "possible_neighbor_gene_or_background",
TRUE ~ "ambiguous"
)
) %>%
left_join(candidate, by = c("locus_id", "celltype_safe")) %>%
arrange(celltype, locus_raw, group)
summary_de <- summary %>%
filter(!is.na(avg_log2FC))
write.csv(summary_de, file.path(res_dir, paste0("hERV_coverage_summary_DE_only_", mode, ".csv")), row.names = FALSE)
ad_summary <- summary %>%
filter(group == "AD") %>%
select(
celltype, locus_raw, locus_id, direction, avg_log2FC, p_val_adj, AD_pct, NC_pct,
assumed_bulk_sc_opposite, coverage_class,
total_herv_raw, total_region_raw,
approx_5p_700bp, body_rest, upstream_5p_10kb, downstream_3p_10kb,
fivep_to_body_density_ratio, downstream_to_herv_density_ratio,
chr, start, end, strand
)
write.csv(ad_summary, file.path(res_dir, paste0("hERV_coverage_AD_focused_DE_only_", mode, ".csv")), row.names = FALSE)
-
hERV_coverage_summary_DE_only_ss_avg.xlsx:预览 | 下载 -
hERV_coverage_AD_focused_DE_only_ss_avg.xlsx:预览 | 下载
| 覆盖类型 | 数量 | 初步解释 |
|---|---|---|
HERV_body_covered |
73 | reads覆盖HERV主体,更多像较长HERV内部转录信号 |
5p_limited_short_like |
18 | reads偏集中在HERV 5’端近似区域,更像短LTR/eRNA-like信号 |
extended_to_3p_flank |
2 | reads向HERV外侧延伸,值得进一步画图和查junction |
reads_too_few |
0 | 有效DE候选里没有reads过少的情况 |
- 差异HERV位点更多覆盖HERV主体,真正明显向外延伸的位点很少
短LTR/eRNA-like候选:
| 细胞类型 | 位点 | 分类 | 方向 |
|---|---|---|---|
| Excit | HERV4_4q22.1 |
5p_limited_short_like |
AD下调 |
| Excit | HERVH_12q13.2b |
5p_limited_short_like |
AD下调 |
| Oligo | HERVL40_15q21.3 |
5p_limited_short_like |
AD下调 |
| Mic | HERVH_2p14b |
5p_limited_short_like |
AD下调 |
向外延伸候选:
| 细胞类型 | 位点 | 分类 | 方向 |
|---|---|---|---|
| Inhib | HML3_5p15.33d |
extended_to_3p_flank |
AD下调 |
| Oligo | MER61_21q21.1b |
extended_to_3p_flank |
AD下调 |
HERV主体覆盖候选:
| 细胞类型 | 位点 | 分类 | 方向 |
|---|---|---|---|
| Oligo | HML2_3q12.3 |
HERV_body_covered |
AD下调 |
| Excit | ERVLE_3q13.33c |
HERV_body_covered |
AD下调 |
| Excit | HML4_16p13.3 |
HERV_body_covered |
AD下调 |
4. IGV看BAM
- 因为看的是hERV,所以在导入基因gtf的基础上还要导入
herv_locus.bed
以Oligo MER61-21q21.1b为例:
-
Load from File- all.Oligo.AD.ss_avg.bw
- all.Oligo.NC.ss_avg.bw
- 可选:all.Oligo.AD.ss_avg.bam
- 可选:all.Oligo.NC.ss_avg.bam
- 跳转到HERV附近区域:在IGV顶部搜索框输入
chr21:20728070-20831214(MER61_21q21.1b上下游约50kb),如果想看更细,输入chr21:20773070-20786214(上下游约5kb) - 调整显示方式:
- 右键点击AD和NC的BigWig轨道,选择
Set Data Range...,把两个轨道设成相同范围,比如0到同一个最大值。否则IGV自动缩放会让AD和NC看起来差不多高,影响比较 - 右键点击BAM轨道,可以选择
Expanded,这样能看到单条read堆积。如果reads太多,可以用Squished
如果BAM轨道中有带N的CIGAR,IGV会显示跨内含子的spliced alignment
- 右键点击AD和NC的BigWig轨道,选择
-
基因注释轨道/hERV BED轨道里的
<:基因/HERV位点在负链read/alignment里的小箭头或方向提示:表示read比对到参考基因组的方向。对于10x/snRNA-seq数据,这个方向不一定能直接等同于真实转录方向,尤其是Stellarscope重分配后的HERV reads
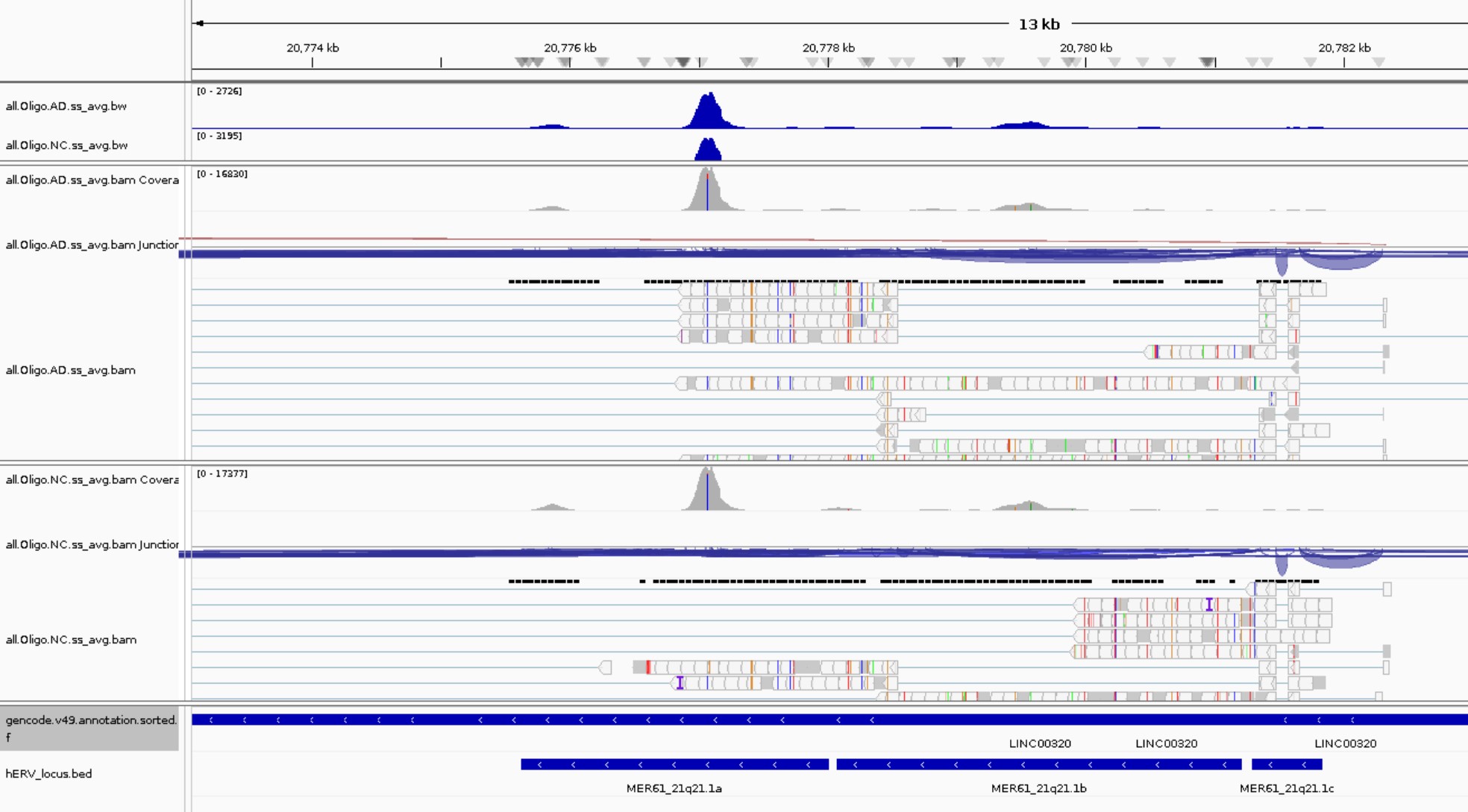
-
all.oligo.AD_ss_avg.bw和all.oligo.NC_ss_avg.bw:stellarscope-updated.bam生成的BigWig覆盖轨道。蓝色峰越高,表示这个位置附近被Stellarscope分配后的reads/alignment覆盖越多注意左侧标尺y轴范围不同,右键轨道→
Set Data Range...可把两个BigWig统一到相同范围,比如都设成0-3000 -
BAM Coverage:BAM文件自带的coverage轨道,灰色柱子也是覆盖深度,和BigWig类似,但来自BAM实时计算,彩色小竖线通常表示该位置的reads碱基与参考基因组不一致,主要是mismatch显示,不是新的表达峰 -
BAM Junction:蓝色弧线是RNA-seq中跨剪接位点的reads,弧线连接的两端是read比对到的两个外显子片段,中间跳过的是内含子或被spliced out的区域 -
BAM reads:真实alignment,灰色小块是read实际比对上的片段,中间细线连接的是spliced alignment的不同片段,彩色竖线同样多是mismatch
补上了STAR的bam:
- all.Oligo.AD.star.bw
- all.Oligo.NC.star.bw
- all.Oligo.AD.star.bam
- all.Oligo.NC.star.bam
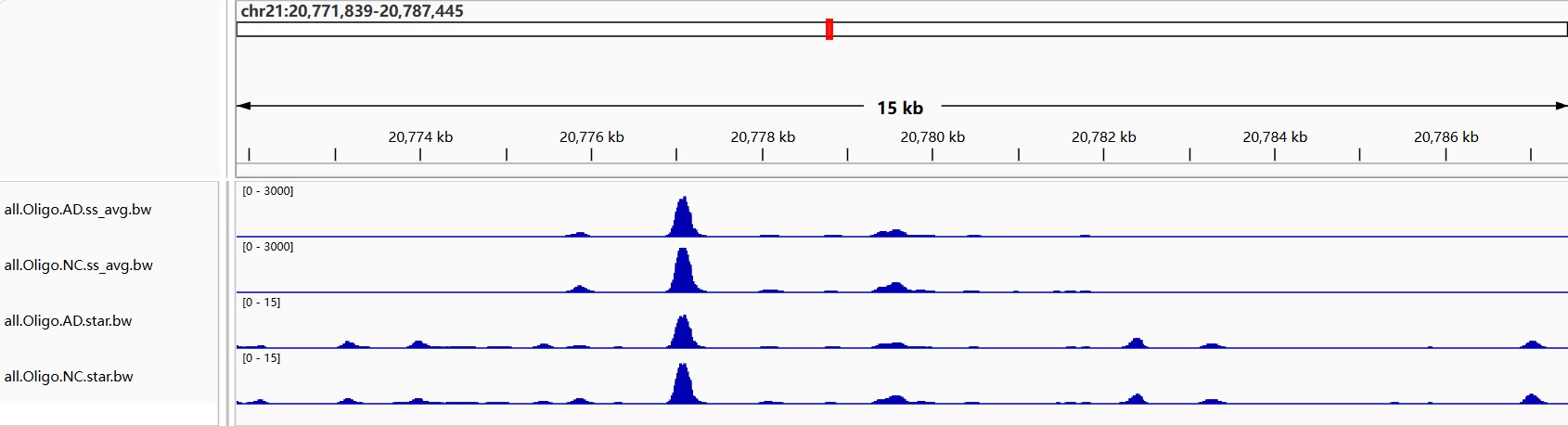

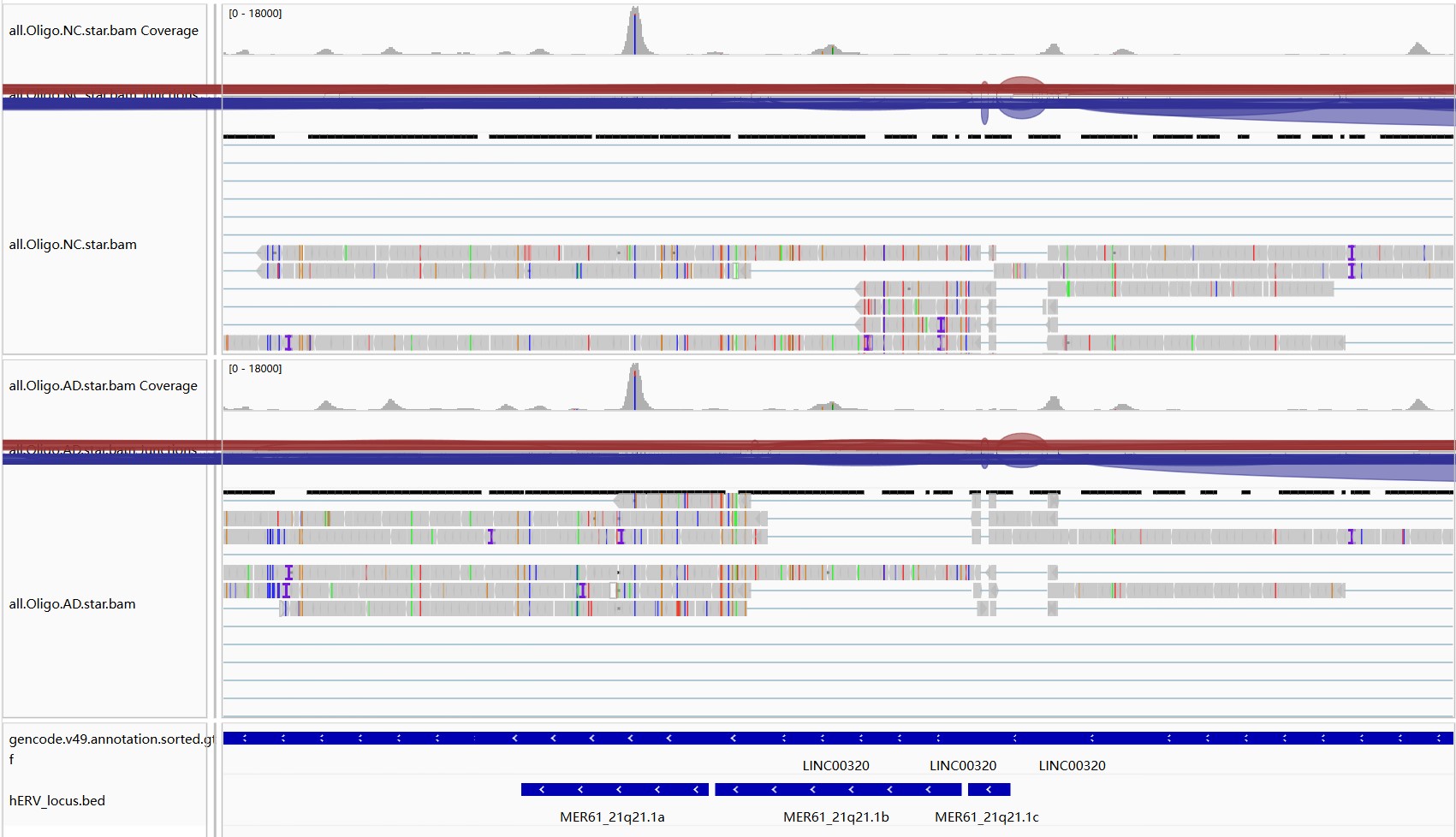
5. pyGenomeTracks看BW
if [[ $# -lt 6 ]]; then
echo "Usage: bash $0 <celltype> <locus> <chr> <start> <end> <strand> [range]" >&2
echo "Example: bash $0 Oligo MER61_21q21.1b chr21 20778070 20781214 - 5000" >&2
exit 1
fi
celltype=$1
locus=$2
chr=$3
start=$4
end=$5
strand=$6
range=${7:-5000}
res_dir=${RES_DIR:-/public/home/GENE_proc/wth/GSE174367/0530test/hERV_coverage}
gtf=${GTF:-/public/home/wangtianhao/Desktop/STAR_ref/gencode.v49.annotation.gtf}
all_herv_bed=${HERV_BED:-/public/home/GENE_proc/wth/GSE174367/0530test/data/hERV_locus.bed}
out_root=${OUT_ROOT:-${res_dir}/track_plots}
five_len=${FIVE_LEN:-700}
flank_len=${FLANK_LEN:-10000}
ymax=${YMAX:-3500}
diff_ymax=${DIFF_YMAX:-1500}
threads=${THREADS:-2}
safe_locus=$(echo "${locus}" | sed 's#[/: ]#_#g')
plot_dir=${out_root}/${safe_locus}_${celltype}_${range}bp
mkdir -p "${plot_dir}"
region_start=$((start - range))
if [[ ${region_start} -lt 0 ]]; then region_start=0; fi
region_end=$((end + range))
region=${chr}:${region_start}-${region_end}
ad_bw=${res_dir}/bigwig_ss_avg/all.${celltype}.AD.ss_avg.bw
nc_bw=${res_dir}/bigwig_ss_avg/all.${celltype}.NC.ss_avg.bw
for f in "${ad_bw}" "${nc_bw}" "${gtf}" "${all_herv_bed}"; do
if [[ ! -f "${f}" ]]; then
echo "[ERROR] File not found: ${f}" >&2
exit 1
fi
done
if [[ ${strand} == "+" ]]; then
five_start=${start}
five_end=$((start + five_len))
if [[ ${five_end} -gt ${end} ]]; then five_end=${end}; fi
flank_start=${end}
flank_end=$((end + flank_len))
elif [[ ${strand} == "-" ]]; then
five_start=$((end - five_len))
if [[ ${five_start} -lt ${start} ]]; then five_start=${start}; fi
five_end=${end}
flank_start=$((start - flank_len))
if [[ ${flank_start} -lt 0 ]]; then flank_start=0; fi
flank_end=${start}
else
echo "[ERROR] strand must be + or -" >&2
exit 1
fi
echo -e "${chr}\t${start}\t${end}\t${locus}\t0\t${strand}" > ${plot_dir}/${safe_locus}.body.bed
echo -e "${chr}\t${five_start}\t${five_end}\tapprox_5p_${five_len}bp\t0\t${strand}" > ${plot_dir}/${safe_locus}.approx5p.bed
echo -e "${chr}\t${flank_start}\t${flank_end}\t3p_flank_${flank_len}bp\t0\t${strand}" > ${plot_dir}/${safe_locus}.3p_flank.bed
awk -v c="${chr}" -v s=${region_start} -v e=${region_end} 'BEGIN{OFS="\t"} $1==c && $2<e && $3>s {print}' \
"${all_herv_bed}" > ${plot_dir}/nearby_hERV.bed
awk -v c="${chr}" -v s=${region_start} -v e=${region_end} 'BEGIN{OFS="\t"}
$1==c && $4<=e && $5>=s && $3=="gene" {
name=$0
if(name ~ /gene_name "/){sub(/.*gene_name "/, "", name); sub(/".*/, "", name)} else {name=$9}
print $1, $4-1, $5, name, 0, $7
}' "${gtf}" > ${plot_dir}/genes.span.bed
awk -v c="${chr}" -v s=${region_start} -v e=${region_end} 'BEGIN{OFS="\t"}
$1==c && $4<=e && $5>=s && $3=="exon" {
name=$0
if(name ~ /gene_name "/){sub(/.*gene_name "/, "", name); sub(/".*/, "", name)} else {name=$9}
print $1, $4-1, $5, name, 0, $7
}' "${gtf}" > ${plot_dir}/genes.exon.bed
awk -v c="${chr}" -v s=${region_start} -v e=${region_end} 'BEGIN{OFS="\t"}
$1==c && $4<=e && $5>=s && $3=="gene" {
name=$0
if(name ~ /gene_name "/){sub(/.*gene_name "/, "", name); sub(/".*/, "", name)} else {name=$9}
gs=$4-1; ge=$5; mid=int((gs+ge)/2); label_pos=mid
if(label_pos<s || label_pos>e){label_pos=int((s+e)/2)}
label_start=label_pos-100; if(label_start<0){label_start=0}
print c, label_start, label_pos+100, name, 0, $7
}' "${gtf}" > ${plot_dir}/genes.label.bed
module load miniconda3/base
conda activate hERV_coverage
bigwigCompare \
-b1 "${ad_bw}" \
-b2 "${nc_bw}" \
--operation subtract \
--binSize 10 \
--region ${chr}:${region_start}:${region_end} \
-p ${threads} \
-o ${plot_dir}/AD_minus_NC.bw
cat > ${plot_dir}/${celltype}.${safe_locus}.tracks.ini <<EOF_INI
[x-axis]
where = top
[${celltype} AD]
file = ${ad_bw}
title = AD
height = 2.5
color = #2ca02c
min_value = 0
max_value = ${ymax}
number_of_bins = 800
nans_to_zeros = true
[${celltype} NC]
file = ${nc_bw}
title = NC
height = 2.5
color = #2ca02c
min_value = 0
max_value = ${ymax}
number_of_bins = 800
nans_to_zeros = true
[AD minus NC]
file = ${plot_dir}/AD_minus_NC.bw
title = AD-NC
height = 2
color = #555555
min_value = -${diff_ymax}
max_value = ${diff_ymax}
number_of_bins = 800
nans_to_zeros = true
grid = true
[hERV body]
file = ${plot_dir}/${safe_locus}.body.bed
title = hERV body
height = 0.6
display = collapsed
color = red
labels = true
[approx 5p]
file = ${plot_dir}/${safe_locus}.approx5p.bed
title = approx 5'
height = 0.6
display = collapsed
color = orange
labels = true
[3p flank]
file = ${plot_dir}/${safe_locus}.3p_flank.bed
title = 3' flank
height = 0.6
display = collapsed
color = blue
labels = true
[nearby hERVs]
file = ${plot_dir}/nearby_hERV.bed
title = nearby hERVs
height = 1.2
display = stacked
color = grey
labels = true
[gene span]
file = ${plot_dir}/genes.span.bed
title = gene span
height = 0.8
display = collapsed
labels = false
color = lightgrey
[gene label]
file = ${plot_dir}/genes.label.bed
title = gene label
height = 0.8
display = collapsed
labels = true
fontsize = 10
color = white
[exons]
file = ${plot_dir}/genes.exon.bed
title = exons
height = 2
display = stacked
labels = false
color = black
EOF_INI
conda activate pygenometracks
pyGenomeTracks \
--tracks ${plot_dir}/${celltype}.${safe_locus}.tracks.ini \
--region ${region} \
--outFileName ${plot_dir}/${celltype}.${safe_locus}.coverage.${range}bp.pdf \
--width 38 \
--height 16 \
--dpi 300 \
--trackLabelFraction 0.22
echo "[DONE] ${plot_dir}/${celltype}.${safe_locus}.coverage.${range}bp.pdf"
bash plot_hERV_bw.sh Oligo MER61_21q21.1b chr21 20778070 20781214 - 5000
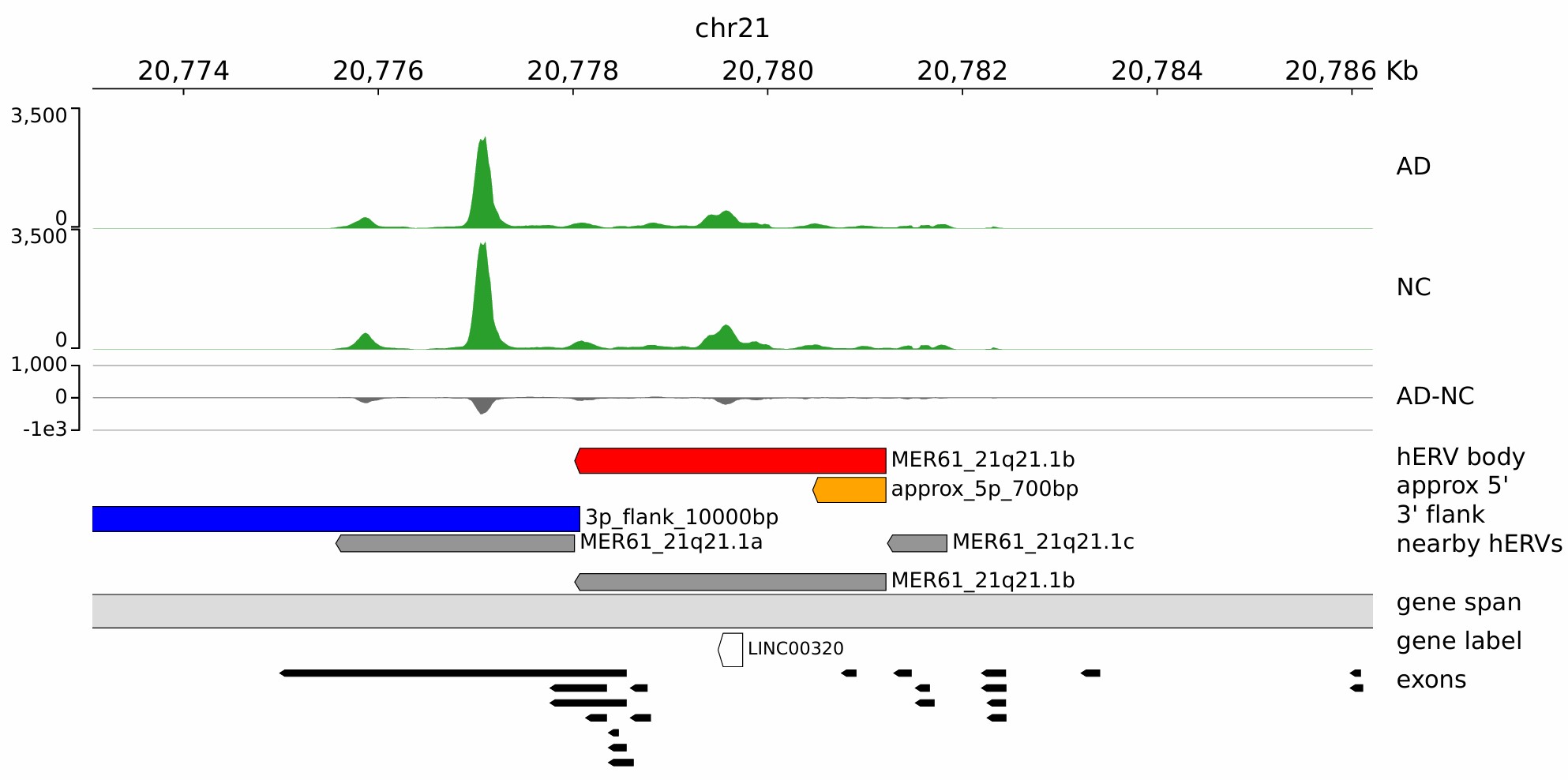
- 红色:MER61_21q21.1b本体
- gene label显示为LINC00320,覆盖整个区域,说明这个HERV位点处在一个lncRNA基因区域内
-
橙色:approx5p(approximate 5’ end),近似5’端区域。因为这里的hERV注释没有给出5’LTR,所以根据HERV方向,把HERV转录方向最前面的700bp当作近似5’端
真正严格的5’LTR应该来自RepeatMasker子结构注释,或者专门的HERV结构注释,能区分5’LTR/internal/3’LTR,这里仅作转录方向的示意
- 蓝色:3p flank,HERV转录方向3’端外侧的邻近区域(HERV结束以后,沿转录方向继续往外的一段区域),用来判断reads是否从HERV向外延伸
- reads主要集中在approx5p:5’端局限的短RNA-like信号或LTR局部启动/增强子样转录,偏向短LTR/eRNA-like覆盖模式
- reads覆盖整个hERV body(覆盖红色HERV主体):不是单纯5’端小峰,更可能是HERV内部较长转录/完整或部分HERV转录本/HERV位于宿主转录本内部,被宿主转录带出来
-
reads明显进入3p flank(reads从HERV本体继续延伸到蓝色3p flank):不是短信号,而是更长的HERV相关转录本/HERV参与的lncRNA-like转录/HERV与邻近基因或lncRNA相关的转录
注:3p flank有覆盖,不等于一定是HERV产生的长lncRNA,也可能是邻近基因或lncRNA本身的reads/附近其他HERV或重复元件的reads/多重比对后分配造成的局部堆积
- 在这张图中,HERV本体附近有信号,同时左侧3’flank附近有明显覆盖峰,因此不太像 “单纯5’LTR上的短信号”,更像“外侧区域有较强覆盖”。但由于该区域位于LINC00320内,不能仅凭coverage说它一定是HERV产生的长lncRNA,也可能与邻近lncRNA转录有关
reads读数是否考虑样本数影响:
-
BigWig部分考虑了测序深度/细胞数影响
scale=$(awk -v n=${total} 'BEGIN{printf "%.10f", 1000000/n}') bamCoverage --bam ${out_bam} -o ${out_bw} -of bigwig --binSize 10 --normalizeUsing None --scaleFactor ${scale} BigWig值 ≈ 当前bin覆盖reads数 × 1,000,000 / 该celltype×group的总assigned alignments数不过这种并没有考虑样本数的影响(reads多/细胞多的样本贡献更大),当然这个其实可以忽略掉,之前snRNA-seq分析时也没看哪个样本中哪类细胞多
-
IGV里的BAM Coverage轨道没有归一化,是IGV直接从BAM算出来的原始coverage。因此主要用来看“reads落在哪里”以及跨剪接、峰是否集中在hERV端、是否向外延伸,而不能直接判断AD-NC的差异
//todo 如果像incRNA就像基因可以看受哪些元件调控
snATAC-seq
利用《Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease》中提供的GSE174367数据集里面的snATAC-seq数据,试一试基本的表观遗传学分析
- 表观遗传计数方法没有和hERV计数方法一样有telescope这种EM特殊方法?先仿照作者流程或按照常规分析流程
- 看看作者是否提供了整理好的表观遗传数据
基础概念和原文思路
ATAC-seq:
- 测的是什么:细胞核里的DNA通常被核小体和染色质结构包裹。某些区域比较开放,转录因子、RNA聚合酶等更容易结合,常见于promoter/enhancer/insulator/TSS或其它调控元件附近、某些活跃的重复序列/LTR区域
- 方法:ATAC-seq使用一种叫Tn5 transposase的酶。这个酶更容易进入开放染色质区域,并在那里切割DNA、接上测序接头。后续测序得到的reads越多,说明该区域越开放
-
peak:reads会集中落在一些开放区域。把这些reads富集区域识别出来,就叫peak,写成
chr19:44904000-44905000这种基因组坐标的形式peak calling就是指从大量ATAC片段中识别“开放区域”
- 数据类型:在snATAC-seq中是peak×cell矩阵(例如行名是基因组坐标,列名是细胞名),数值表示该细胞在这个peak里检测到多少ATAC片段。因为单细胞/单核ATAC数据很稀疏,很多时候也可以近似理解为“这个peak在这个细胞里有没有被检测到”
- 为什么说不用EM这类特殊方法可能会对结果有影响:默认ATAC流程会过滤多重比对reads,所以对高度重复的hERV区域偏保守,可能低估开放性
原文中作者是怎么分析的:
-
比对计数:
cellranger-atac count(v1.1.0, 10X Genomics),具体参数和方法未说明 -
基础snATAC分析:snATAC矩阵先经过latent semantic indexing(LSI)降维,然后用mutual nearest neighbors(MNN)校正批次效应;之后做UMAP和Leiden聚类,并根据已知marker gene启动子/基因区域附近的开放性注释出主要脑细胞类型
-
与snRNA整合:把snATAC中的gene activity作为query,把snRNA中的gene expression作为reference,用Seurat的整合框架找anchors,并用label transfer验证snATAC细胞类型注释 -> 大多数snATAC细胞都能得到较高的预测分数,说明snRNA和snATAC两种数据的细胞类型对应关系较好
- 作者想验证snATAC-seq区分的细胞类型是否准确,因为snRNA-seq的表达量在细胞类型注释中是最准确的,所以就要用snRNA-seq作为参照。简单来说,就是用Seurat的anchor方法,比如snRNA里已经注释出一群小胶质细胞,snATAC里也有一群细胞在P2RY12、CSF1R、CX3CR1等小胶质相关基因附近很开放,那么Seurat会认为它们在共同空间里很接近,于是建立对应关系
- 不是说同一个细胞同时测了RNA和ATAC,而是说“这两个细胞虽然来自不同测序实验,但它们的分子特征很像,可能属于同一种细胞类型”
- gene activity:如果某个基因的启动子、基因体或附近调控区域在某个snATAC细胞中很开放,就认为这个基因在这个细胞中具有较高gene activity,不是具体表达量,而只是“这个基因附近的染色质是否开放”的估计值。因为peak×cell矩阵不能直接和gene×cell矩阵比较,所以作者做了这个类似矩阵
- label transfer:把reference里的标签转移给query。比如snRNA-seq中某个cluster已经标注为microglia,Seurat发现snATAC中某些细胞和这群snRNA microglia很接近,就把microglia这个标签预测给这些snATAC细胞
- 在大细胞类型内部又重新做UMAP和Leiden聚类,识别细胞亚群,也是用snRNA聚类作为reference做label transfer。例如在少突胶质细胞中,作者根据gene activity和gene expression把细胞状态分成progenitor、intermediate、mature等阶段
- 在每个细胞cluster中比较late-stageAD和control,找differentially accessible chromatin regions(AD相关差异开放区域) -> 差异开放区域和差异表达基因都有很强的亚群特异性;这些区域附近基因的GO富集也呈现明显细胞类型特异性
- 转录因子分析:用chromVAR分析TF motif活性(先判断每个peak里是否含有某个TF motif,再计算每个单核中这些motif所在peak的开放性偏离程度) -> AD中某些TF motif变化具有细胞类型特异性
- 在每个细胞类型的亚群中call peak,发现细胞类型特异的调控区域
- 用Cicero构建cis co-accessibility networks(CCANs):如果两个peak在一群细胞中经常一起开放,它们可能处在同一个调控单元中(peak B可能和peak A所在基因存在调控关系)
co-accessibility能帮助找enhancer-gene关系:很多enhancer离它调控的基因很远,不一定是最近的那个基因。作者就是先构建cis co-accessibility networks,也就是在局部基因组范围内找共同开放的peak。然后重点关注“一个peak落在promoter、另一个peak是远端开放区域”的peak pair。随后作者又用snRNA表达数据检查“这个候选基因的表达是否和该cCRE的开放性相关”,得到gene-linked candidate cis-regulatory elements(gl-cCREs)
- 用NMF把gl-cCRE按照开放模式分成30个模块,判断每个模块对应哪个细胞类型或AD/control状态,并对这些模块靶基因做GO富集。发现一个少突胶质细胞相关调控模块,其靶基因包括APOE和CLU,这两个都是AD相关基因
-
把snATAC开放区域和ADGWAS风险位点结合起来
- 用LDSC看不同cluster的特异开放peak是否富集某些复杂性状/疾病的遗传力:peak向上下游各扩展2000bp,要求在该cluster至少1%细胞中开放,并去除在多个细胞类型中也开放的peak;然后用LDSC做分区遗传力富集
- 在具体AD风险位点上,把co-accessibility网络、开放性信号和GWAS统计量放到同一个基因组坐标轴上,分析可能被风险变异影响的调控关系
-
与snRNA整合:把snATAC中的gene activity作为query,把snRNA中的gene expression作为reference,用Seurat的整合框架找anchors,并用label transfer验证snATAC细胞类型注释 -> 大多数snATAC细胞都能得到较高的预测分数,说明snRNA和snATAC两种数据的细胞类型对应关系较好
原文snATAC部分的主要结论:
- AD脑组织中存在细胞类型特异的染色质开放变化
- 小胶质细胞、星形胶质细胞和少突胶质细胞是重点变化细胞类型,其中小胶质细胞激活状态、反应性星形胶质细胞状态、免疫相关少突胶质状态都与AD有关
- snATAC和snRNA整合可以把开放调控元件连接到靶基因,作者得到大量gl-cCRE-gene关系,并发现与APOE、CLU等AD相关基因有关的少突胶质细胞调控模块
- TF层面发现多个疾病相关调控因子,例如小胶质细胞中的SPI1/PU.1、少突胶质细胞中的NRF1/SREBF1相关信号
- ADGWAS风险位点可以被解释到具体细胞类型和调控区域中,例如APOE位点在小胶质细胞和星形胶质细胞中存在疾病相关调控网络改变
尝试分析
library(Seurat)
library(Signac)
library(edgeR)
library(tidyverse)
library(Matrix)
library(patchwork)
library(openxlsx)
library(readxl)
library(GenomicRanges)
library(rtracklayer)
res_dir <- "/public/home/GENE_proc/wth/GSE174367/0530test/"
数据预处理
# 读取数据
peak_counts <- Read10X_h5("/public/home/GENE_proc/wth/GSE174367/data_snATAC/GSE174367_snATAC-seq_filtered_peak_bc_matrix.h5")
meta <- read.csv("/public/home/GENE_proc/wth/GSE174367/data_snATAC/GSE174367_snATAC-seq_cell_meta.csv")
rownames(meta) <- meta$Barcode
common_cells <- intersect(colnames(peak_counts), rownames(meta))
peak_counts <- peak_counts[, common_cells]
meta <- meta[common_cells, , drop = FALSE]
> dim(peak_counts)
[1] 219070 130418
# 创建Signac对象
chrom_assay <- CreateChromatinAssay(
counts = peak_counts,
sep = c(":", "-"),
genome = "hg38",
min.cells = 10
)
atac <- CreateSeuratObject(
counts = chrom_assay,
assay = "peaks",
meta.data = meta,
project = "GSE174367_snATAC"
)
colnames(atac@meta.data) <- c("dataset", "nCount_peaks", "nFeature_peaks", "sample_id", "batch", "age", "sex", "PMI", "tangle_Stage", "plaque_Stage", "group", "RIN", "cluster", "celltype", "barcode")
> table(atac$sample_id)
Sample-100 Sample-101 Sample-17 Sample-19 Sample-22 Sample-27 Sample-33 Sample-37 Sample-40 Sample-43 Sample-45 Sample-46 Sample-47
4315 9787 8822 3466 4568 2605 3197 5272 4889 3076 6879 9488 10048
Sample-50 Sample-52 Sample-58 Sample-66 Sample-82 Sample-90 Sample-96
4139 9403 7881 6317 8363 5932 11971
> table(atac$group)
AD Control
66449 63969
> table(atac$celltype)
ASC EX INH MG ODC OPC PER.END
15399 24076 9644 12232 62253 4869 1945
> table(atac$cluster)
ASC.a ASC.b ASC.c ASC.d ASC.e ASC.f EX.a EX.b EX.c EX.d EX.e INH.a INH.b INH.c INH.d
6062 4215 2870 1148 828 276 6356 6065 5706 4590 1359 4088 3914 1418 224
MG.a MG.b MG.c MG.d MG.e ODC.a ODC.b ODC.c ODC.d ODC.e ODC.f ODC.g ODC.h ODC.i ODC.j
6439 4352 792 475 174 6459 6248 6148 5956 5578 5288 4906 4732 3755 3610
ODC.k ODC.l ODC.m OPC.a PER.END.a
3480 3325 2768 4869 1945
# 基础QC可视化
p1 <- VlnPlot(
atac,
features = c("nCount_peaks", "nFeature_peaks"),
group.by = "group",
pt.size = 0,
ncol = 2
)
p2 <- VlnPlot(
atac,
features = c("nCount_peaks", "nFeature_peaks"),
group.by = "sample_id",
pt.size = 0,
ncol = 1
)
p1 / p2
# TF-IDF、LSI降维
DefaultAssay(atac) <- "peaks"
atac <- RunTFIDF(atac)
atac <- FindTopFeatures(atac, min.cutoff = "q0")
atac <- RunSVD(atac)
DepthCor(atac)
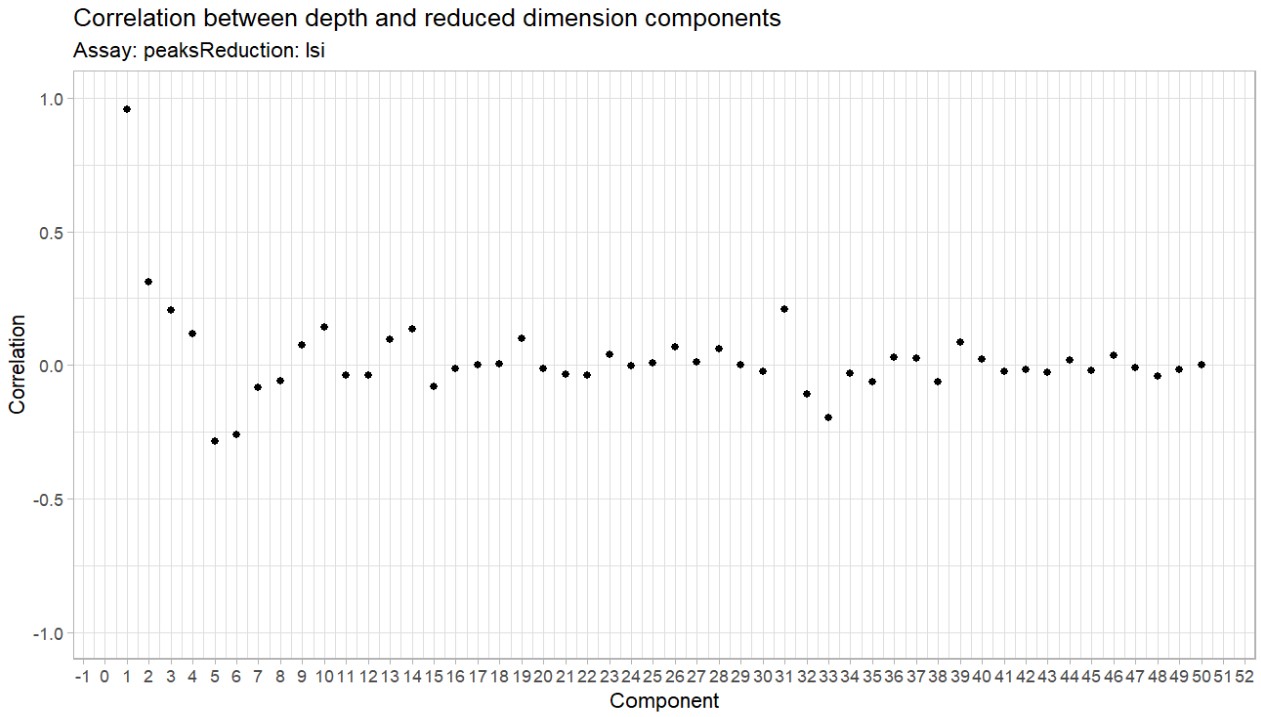
- 横轴是LSI降维后的component(可以类比RNA-seq里的PC1、PC2、PC3),纵轴是每个component和测序深度的相关性
- 检查每个LSI维度是不是主要反映测序深度,而不是生物学差异:在snATAC-seq中,每个细胞核检测到的fragments/peaks数量差别很大。有些细胞测序深度高,就会看到更多开放区域;有些细胞测序深度低,就会看到更少开放区域。这个差异可能只是技术因素,不是真正的细胞类型差异
- 可以看到Component 1和depth的相关性接近1,这说明LSI第1维几乎完全由测序深度驱动,所以后面不应该用第1维做UMAP/聚类
# UMAP聚类
dims_use <- 2:30 # 通常LSI第1维和测序深度关系比较强
atac <- RunUMAP(
atac,
reduction = "lsi",
dims = dims_use
)
atac <- FindNeighbors(
atac,
reduction = "lsi",
dims = dims_use
)
atac <- FindClusters(
atac,
resolution = 0.3
)
p_cluster <- DimPlot(
atac,
reduction = "umap",
group.by = "seurat_clusters",
label = TRUE
) + ggtitle("snATAC clusters")
p_group <- DimPlot(
atac,
reduction = "umap",
group.by = "group"
) + ggtitle("Diagnosis")
p_sample <- DimPlot(
atac,
reduction = "umap",
group.by = "sample_id"
) + ggtitle("Sample")
# 用作者注释出的细胞类型
p_celltype <- DimPlot(
atac,
reduction = "umap",
group.by = "celltype",
label = TRUE,
repel = TRUE
) + ggtitle("Author cell type")
(p_cluster | p_group) / (p_sample | p_celltype)
save(atac, file = file.path(res_dir, "atac_clustered.RData"))
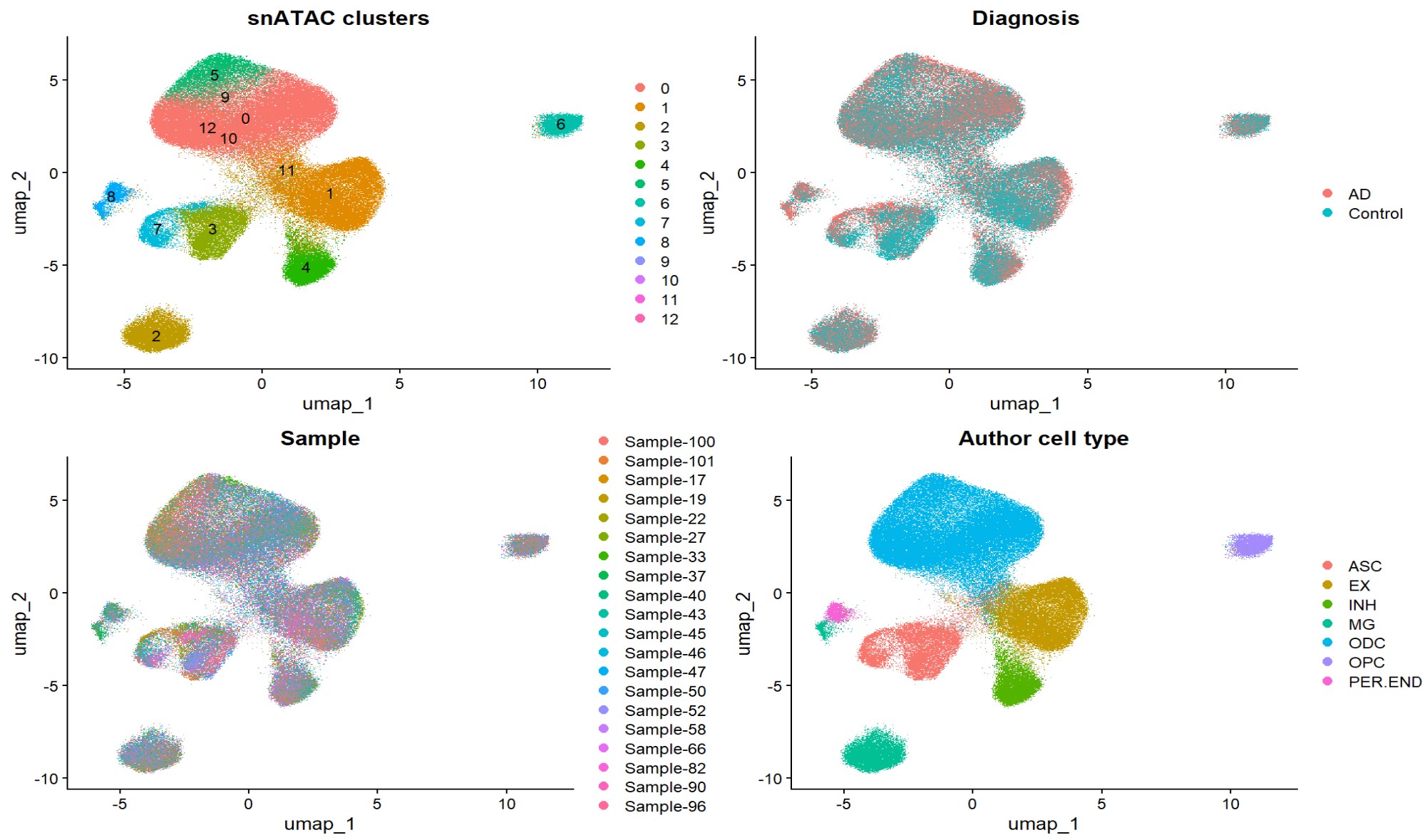
> table(atac$seurat_clusters, atac$celltype)
ASC EX INH MG ODC OPC PER.END
0 303 69 34 220 51356 29 22
1 1275 23414 994 561 2531 146 142
2 63 94 19 10346 164 4 21
3 9734 22 7 70 22 4 2
4 65 389 8573 36 56 6 3
5 33 0 0 28 7900 3 0
6 65 71 10 60 183 4672 0
7 3764 1 0 14 3 1 1
8 96 16 7 896 32 4 1754
9 0 0 0 0 2 0 0
10 0 0 0 0 2 0 0
11 1 0 0 0 1 0 0
12 0 0 0 1 1 0 0
这个结果大致可以对应上作者的细胞类型,后续就直接用作者的细胞类型了
按细胞类型找差异开放peak(差异可及区域, DAR)
pseudobulk:
atac$group <- factor(atac$group, levels = c("Control", "AD"))
# 每个细胞类型中每个样本的细胞数
celltype_sample_n <- atac@meta.data %>%
as.data.frame() %>%
count(celltype, sample_id, group, name = "n_cells") %>%
arrange(celltype, group, sample_id)
# pseudobulk差异开放分析
counts_peak <- GetAssayData(atac, assay = "peaks", layer = "counts")
celltypes <- c("ASC", "EX", "INH", "MG", "ODC", "OPC", "PER.END")
min_cells_per_sample <- 50
all_res <- list()
summary_res <- list()
for (ct in celltypes) {
message("Running celltype: ", ct)
md_ct <- atac@meta.data %>%
as.data.frame() %>%
rownames_to_column("cell") %>%
filter(celltype == ct)
sample_keep <- md_ct %>%
count(sample_id, group, name = "n_cells") %>%
filter(n_cells >= min_cells_per_sample)
md_ct <- md_ct %>%
filter(sample_id %in% sample_keep$sample_id)
n_ad <- length(unique(md_ct$sample_id[md_ct$group == "AD"]))
n_ctrl <- length(unique(md_ct$sample_id[md_ct$group == "Control"]))
if (n_ad < 2 | n_ctrl < 2) {
message("Skip ", ct, ": too few samples after filtering")
next
}
cells_use <- md_ct$cell
X <- counts_peak[, cells_use, drop = FALSE]
sample_factor <- factor(md_ct$sample_id)
M <- Matrix::sparse.model.matrix(~ 0 + sample_factor)
colnames(M) <- levels(sample_factor)
pb_counts <- X %*% M
sample_md <- md_ct %>%
count(sample_id, group, name = "n_cells") %>%
arrange(match(sample_id, colnames(pb_counts)))
rownames(sample_md) <- sample_md$sample_id
sample_md$group <- factor(sample_md$group, levels = c("Control", "AD"))
y <- DGEList(
counts = as.matrix(pb_counts),
samples = sample_md
)
keep_peak <- filterByExpr(y, group = sample_md$group)
n_before <- nrow(y)
y <- y[keep_peak, , keep.lib.sizes = FALSE]
n_after <- nrow(y)
y <- calcNormFactors(y)
design <- model.matrix(~ group, data = sample_md)
y <- estimateDisp(y, design)
fit <- glmQLFit(y, design, robust = TRUE)
qlf <- glmQLFTest(fit, coef = "groupAD")
res <- topTags(qlf, n = Inf)$table %>%
rownames_to_column("peak") %>%
arrange(FDR) %>%
mutate(
celltype = ct,
n_AD_samples = n_ad,
n_Control_samples = n_ctrl,
n_peaks_before_filter = n_before,
n_peaks_after_filter = n_after,
direction = case_when(
FDR < 0.05 & logFC > 0 ~ "AD_more_open",
FDR < 0.05 & logFC < 0 ~ "AD_less_open",
TRUE ~ "not_significant"
)
) %>%
select(celltype, peak, logFC, logCPM, F, PValue, FDR, direction,
n_AD_samples, n_Control_samples,
n_peaks_before_filter, n_peaks_after_filter)
all_res[[ct]] <- res
summary_res[[ct]] <- data.frame(
celltype = ct,
n_AD_samples = n_ad,
n_Control_samples = n_ctrl,
n_peaks_before_filter = n_before,
n_peaks_after_filter = n_after,
n_sig_FDR_0.05 = sum(res$FDR < 0.05),
n_AD_more_open = sum(res$FDR < 0.05 & res$logFC > 0),
n_AD_less_open = sum(res$FDR < 0.05 & res$logFC < 0)
)
}
all_res_df <- bind_rows(all_res)
summary_df <- bind_rows(summary_res)
wb <- createWorkbook()
addWorksheet(wb, "cell_counts")
writeData(wb, "cell_counts", celltype_sample_n)
addWorksheet(wb, "summary")
writeData(wb, "summary", summary_df)
top_peaks <- all_res_df %>%
group_by(celltype) %>%
arrange(FDR, .by_group = TRUE) %>%
slice_head(n = 50) %>%
ungroup()
sig_peaks <- all_res_df %>%
filter(FDR < 0.05) %>%
arrange(celltype, FDR)
sig_peaks2 <- all_res_df %>%
filter(FDR < 0.1) %>%
arrange(celltype, FDR)
addWorksheet(wb, "top50")
writeData(wb, "top50", top_peaks)
addWorksheet(wb, "sig_FDR_0.05")
writeData(wb, "sig_FDR_0.05", sig_peaks)
addWorksheet(wb, "sig_FDR_0.1")
writeData(wb, "sig_FDR_0.1", sig_peaks2)
saveWorkbook(
wb,
file = file.path(res_dir, "DA_peaks_edgeR_summary.xlsx"),
overwrite = TRUE
)
for (ct in names(all_res)) {
sheet_name <- gsub("[\\[\\]\\*\\?/\\\\:]", "_", ct)
sheet_name <- substr(sheet_name, 1, 31)
addWorksheet(wb, sheet_name)
writeData(wb, sheet_name, all_res[[ct]])
}
saveWorkbook(
wb,
file = file.path(res_dir, "DA_peaks_edgeR.xlsx"),
overwrite = TRUE
)
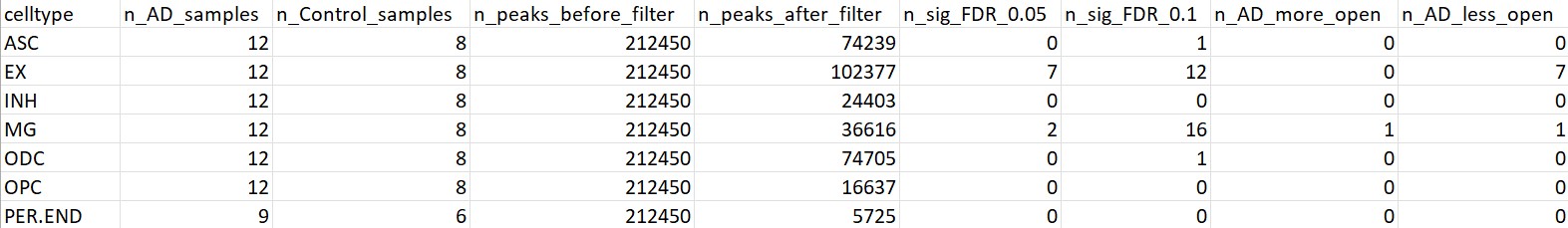
如果将p值阈值设置成0.05的话,可以看到显著结果较少
- 这里的pseudobulk是直接把每个样本的各细胞counts相加
- logFC>0:AD组开放性更高
之后进行亚群-样本级别的分析,结果与上面类似:
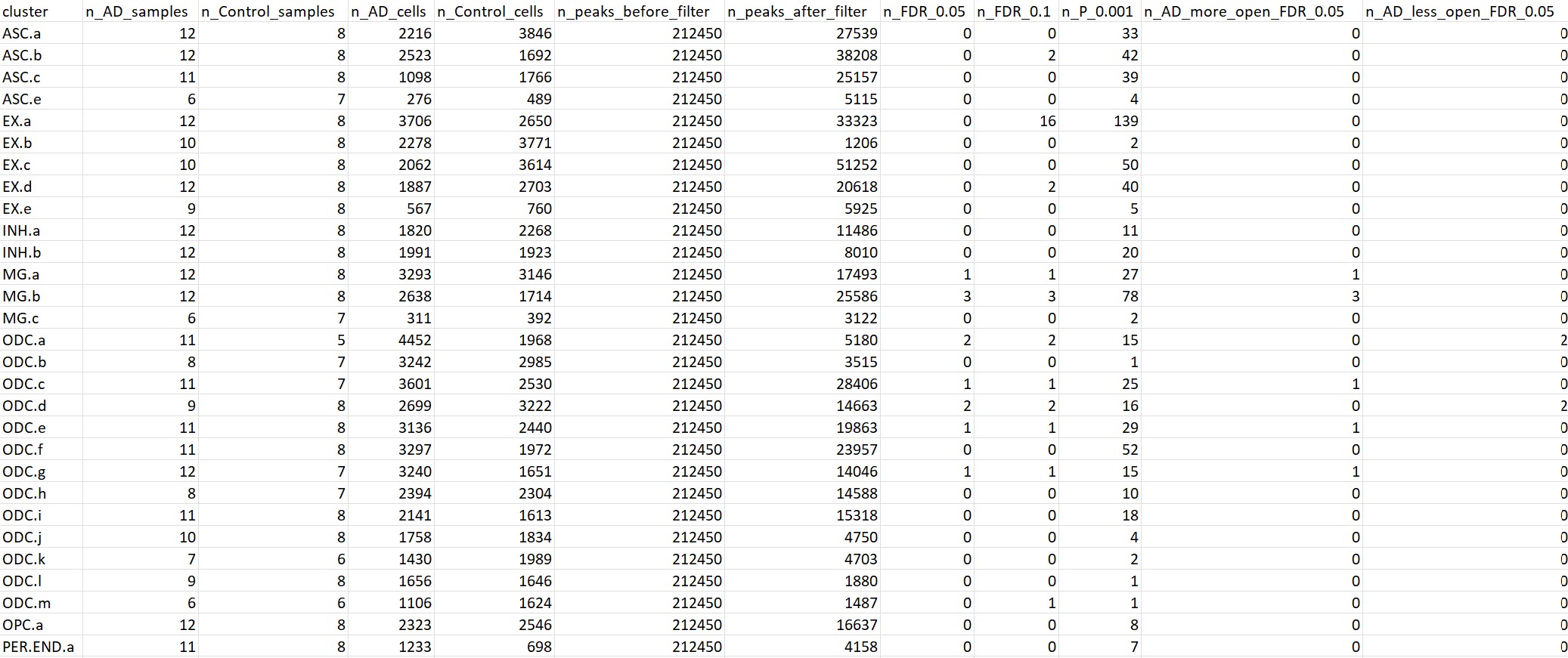
之后看了一下论文中的补充材料,作者并没有用pseudobulk:
- In all cases, we used the Seurat FindAllMarkers function with varying parameters to compare features between groups
- differential chromatin accessibility and differential TF motif variability:used the ‘LR’ option to perform logistic regression analyses
- differential gene expression:used the ‘MAST’ option
# 作者使用的是binarized peak matrix
peak_counts <- GetAssayData(atac, assay = "peaks", layer = "counts")
peak_bin <- peak_counts
peak_bin@x <- rep(1, length(peak_bin@x))
atac[["peaks_bin"]] <- CreateChromatinAssay(
counts = peak_bin,
sep = c("-", "-"),
genome = "hg38",
min.cells = 1
)
DefaultAssay(atac) <- "peaks_bin"
atac$group <- factor(atac$group, levels = c("Control", "AD"))
# DAR
run_dar_lr <- function(seu, subset_col, subset_value) {
md <- seu@meta.data %>%
as.data.frame() %>%
rownames_to_column("cell") %>%
filter(.data[[subset_col]] == subset_value)
n_ad <- sum(md$group == "AD")
n_ctrl <- sum(md$group == "Control")
sub <- subset(seu, cells = md$cell)
DefaultAssay(sub) <- "peaks_bin"
Idents(sub) <- sub[["group", drop = TRUE]]
res <- FindMarkers(
sub,
ident.1 = "AD",
ident.2 = "Control",
assay = "peaks_bin",
test.use = "LR",
only.pos = FALSE,
logfc.threshold = 0,
min.pct = 0.01,
)
res <- res %>%
as.data.frame() %>%
rownames_to_column("peak")
res$p_val_adj_bonf <- p.adjust(res$p_val, method = "bonferroni", n = nrow(seu[["peaks_bin"]]))
fc_col <- intersect(c("avg_log2FC", "avg_logFC"), colnames(res))[1]
res %>%
mutate(
unit = subset_value,
level = subset_col,
n_AD = n_ad,
n_Control = n_ctrl,
direction = case_when(
.data[[fc_col]] > 0 ~ "AD_more_open",
.data[[fc_col]] < 0 ~ "AD_less_open",
TRUE ~ "no_change"
)
) %>%
arrange(p_val_adj_bonf)
}
# 按细胞类型做DAR
celltypes <- sort(unique(atac$celltype))
celltype_res <- list()
for (ct in celltypes) {
message("Running celltype: ", ct)
celltype_res[[ct]] <- run_dar_lr(atac, "celltype", ct)
}
celltype_res <- celltype_res[!sapply(celltype_res, is.null)]
# 按亚群做DAR
clusters <- sort(unique(atac$cluster))
cluster_res <- list()
for (cl in clusters) {
message("Running cluster: ", cl)
cluster_res[[cl]] <- run_dar_lr(atac, "cluster", cl)
}
cluster_res <- cluster_res[!sapply(cluster_res, is.null)]
save(celltype_res, cluster_res, file = file.path(res_dir, "DAR_res.RData"))
# 保存结果
max_rows_per_sheet <- Inf
safe_sheet_name <- function(x) {
x <- gsub("[\\[\\]\\*\\?/\\\\:]", "_", x)
substr(x, 1, 31)
}
# 这里发现FindMarkers要运行很久,就先用作者的结果表格代替
read_author_dar <- function(xlsx_file, level_name) {
df <- read_xlsx(xlsx_file, skip = 2, col_names = FALSE)
df <- df[, 1:7]
colnames(df) <- c("p_val", "avg_logFC", "pct_AD", "pct_Control", "p_val_adj", level_name, "peak")
df %>%
mutate(
p_val = as.numeric(p_val),
avg_logFC = as.numeric(avg_logFC),
pct_AD = as.numeric(pct_AD),
pct_Control = as.numeric(pct_Control),
p_val_adj = as.numeric(p_val_adj),
direction = case_when(
avg_logFC > 0 ~ "AD_more_open",
avg_logFC < 0 ~ "AD_less_open",
TRUE ~ "no_change"
)
) %>%
filter(!is.na(peak), !is.na(p_val))
}
dar_ct <- read_author_dar(
xlsx_file = "C:\\Users\\17185\\Desktop\\file\\ATACtest\\data\\Supplementary Data 5 AD vs Control cell type DARs.xlsx",
level_name = "celltype"
)
dar_cl <- read_author_dar(
xlsx_file = "C:\\Users\\17185\\Desktop\\file\\ATACtest\\data\\Supplementary Data 6 AD vs Control cluster DARs.xlsx",
level_name = "cluster"
)
# 按细胞类型拆分一下
write_split_xlsx <- function(df, split_col, file, na_sheet = "NA") {
x <- as.character(df[[split_col]])
x[is.na(x)] <- na_sheet
sheet_list <- split(df, x, drop = TRUE)
clean_names <- names(sheet_list)
clean_names <- gsub("[\\[\\]\\:\\*\\?\\/\\\\]", "_", clean_names)
clean_names <- trimws(clean_names)
clean_names[clean_names == ""] <- "blank"
clean_names <- substr(clean_names, 1, 31)
clean_names <- make.unique(clean_names, sep = "_")
clean_names <- substr(clean_names, 1, 31)
names(sheet_list) <- clean_names
openxlsx::write.xlsx(sheet_list, file = file, overwrite = TRUE)
}
write_split_xlsx(dar_ct, split_col = "celltype", file = "C:\\Users\\17185\\Desktop\\file\\ATACtest\\data\\celltype_DAR.xlsx")
write_split_xlsx(dar_cl, split_col = "cluster", file = "C:\\Users\\17185\\Desktop\\file\\ATACtest\\data\\cluster_DAR.xlsx")
hERV/LTR overlap
把hERV位点信息转成BED文件:
herv <- read.csv("C:\\Users\\17185\\Desktop\\hERV_AD_snRNA-seq_analyze\\res\\locus_summary\\locus_summary.csv", show_col_types = FALSE)
herv_bed <- herv %>%
mutate(
chrom = str_match(coord, "^([^:]+):(\\d+)-(\\d+)$")[, 2],
start_1based = as.integer(str_match(coord, "^([^:]+):(\\d+)-(\\d+)$")[, 3]),
end = as.integer(str_match(coord, "^([^:]+):(\\d+)-(\\d+)$")[, 4]),
start = start_1based - 1,
name = locus_id,
score = 0
) %>%
select(chrom, start, end, name, score, strand)
write_tsv(
herv_bed,
"C:\\Users\\17185\\Desktop\\file\\ATACtest\\data\\hERV_locus.bed",
col_names = FALSE
)
对细胞类型DAR做overlap:
group_col <- "celltype"
herv_gr <- import("C:\\Users\\17185\\Desktop\\file\\ATACtest\\data\\hERV_locus.bed")
seqlevelsStyle(herv_gr) <- "UCSC"
peak_to_gr <- function(peaks) {
peaks <- as.character(peaks)
if (any(grepl(":", peaks))) {
x <- str_match(peaks, "^(.+):(\\d+)-(\\d+)$")
} else {
x <- str_match(peaks, "^(.+)-(\\d+)-(\\d+)$")
}
gr <- GRanges(
seqnames = x[, 2],
ranges = IRanges(start = as.integer(x[, 3]), end = as.integer(x[, 4]))
)
mcols(gr)$peak <- peaks
gr
}
# 构建扩展范围
herv_gr_2kb <- herv_gr
start(herv_gr_2kb) <- pmax(start(herv_gr_2kb) - 2000, 1)
end(herv_gr_2kb) <- end(herv_gr_2kb) + 2000
herv_gr_10kb <- herv_gr
start(herv_gr_10kb) <- pmax(start(herv_gr_10kb) - 10000, 1)
end(herv_gr_10kb) <- end(herv_gr_10kb) + 10000
# 提取hERV/LTR名称
herv_name <- if ("name" %in% colnames(mcols(herv_gr))) {
mcols(herv_gr)$name
} else if (!is.null(names(herv_gr))) {
names(herv_gr)
} else {
paste0(seqnames(herv_gr), ":", start(herv_gr), "-", end(herv_gr))
}
# overlap注释函数
add_overlap <- function(dar_df, target_gr, prefix) {
peak_gr <- peak_to_gr(dar_df$peak)
ov <- findOverlaps(peak_gr, target_gr, ignore.strand = TRUE)
hit_df <- tibble(
peak = mcols(peak_gr)$peak[queryHits(ov)],
hit_id = herv_name[subjectHits(ov)]
) %>%
group_by(peak) %>%
summarise(hit_id = paste(unique(hit_id), collapse = ";"), .groups = "drop")
dar_df %>%
left_join(hit_df, by = "peak") %>%
mutate(
"{prefix}_overlap" := !is.na(hit_id),
"{prefix}_herv_ltr_id" := hit_id
) %>%
select(-hit_id)
}
dar_anno <- dar_df %>%
add_overlap(herv_gr, "direct") %>%
add_overlap(herv_gr_2kb, "plus2kb") %>%
add_overlap(herv_gr_10kb, "plus10kb") %>%
mutate(
plus2kb_only = plus2kb_overlap & !direct_overlap,
plus10kb_only = plus10kb_overlap & !plus2kb_overlap
)
# 汇总表
summary_df <- dar_anno %>%
group_by(.data[[group_col]]) %>%
summarise(
n_DAR = n(),
n_direct = sum(direct_overlap),
frac_direct = n_direct / n_DAR,
n_plus2kb = sum(plus2kb_overlap),
frac_plus2kb = n_plus2kb / n_DAR,
n_plus10kb = sum(plus10kb_overlap),
frac_plus10kb = n_plus10kb / n_DAR,
n_direct_AD_more_open = sum(direct_overlap & direction == "AD_more_open"),
n_direct_AD_less_open = sum(direct_overlap & direction == "AD_less_open"),
n_plus2kb_AD_more_open = sum(plus2kb_overlap & direction == "AD_more_open"),
n_plus2kb_AD_less_open = sum(plus2kb_overlap & direction == "AD_less_open"),
n_plus10kb_AD_more_open = sum(plus10kb_overlap & direction == "AD_more_open"),
n_plus10kb_AD_less_open = sum(plus10kb_overlap & direction == "AD_less_open"),
.groups = "drop"
) %>%
arrange(desc(n_direct), desc(n_plus2kb), desc(n_plus10kb))
wb <- createWorkbook()
addWorksheet(wb, "summary")
writeData(wb, "summary", summary_df)
addWorksheet(wb, "all_DARs")
writeData(wb, "all_DARs", dar_anno)
addWorksheet(wb, "direct_overlap")
writeData(wb, "direct_overlap", dar_anno %>% filter(direct_overlap) %>% arrange(.data[[group_col]], p_val_adj, p_val))
addWorksheet(wb, "plus2kb_overlap")
writeData(wb, "plus2kb_overlap", dar_anno %>% filter(plus2kb_overlap) %>% arrange(.data[[group_col]], p_val_adj, p_val))
addWorksheet(wb, "plus10kb_overlap")
writeData(wb, "plus10kb_overlap", dar_anno %>% filter(plus10kb_overlap) %>% arrange(.data[[group_col]], p_val_adj, p_val))
saveWorkbook(wb, file = file.path(res_dir, "celltype_DAR_hERV_LTR_overlap_all.xlsx"), overwrite = TRUE)
过滤出显著结果:Bonferroni校正p_val_adj<0.05
group_col <- "celltype"
padj_cutoff <- 0.05
dar <- read_xlsx(file.path(res_dir, "celltype_DAR_hERV_LTR_overlap_all.xlsx"), sheet = "all_DARs") %>%
mutate(
p_val = as.numeric(p_val),
avg_logFC = as.numeric(avg_logFC),
pct_AD = as.numeric(pct_AD),
pct_Control = as.numeric(pct_Control),
p_val_adj = as.numeric(p_val_adj),
direct_overlap = as.logical(direct_overlap),
plus2kb_overlap = as.logical(plus2kb_overlap),
plus10kb_overlap = as.logical(plus10kb_overlap)
)
sig <- dar %>%
filter(p_val_adj < padj_cutoff) %>%
arrange(.data[[group_col]], p_val_adj, p_val)
summary_sig <- sig %>%
group_by(.data[[group_col]]) %>%
summarise(
n_sig_DAR = n(),
n_direct = sum(direct_overlap, na.rm = TRUE),
frac_direct = n_direct / n_sig_DAR,
n_plus2kb = sum(plus2kb_overlap, na.rm = TRUE),
frac_plus2kb = n_plus2kb / n_sig_DAR,
n_plus10kb = sum(plus10kb_overlap, na.rm = TRUE),
frac_plus10kb = n_plus10kb / n_sig_DAR,
n_direct_AD_more_open = sum(direct_overlap & direction == "AD_more_open", na.rm = TRUE),
n_direct_AD_less_open = sum(direct_overlap & direction == "AD_less_open", na.rm = TRUE),
n_plus2kb_AD_more_open = sum(plus2kb_overlap & direction == "AD_more_open", na.rm = TRUE),
n_plus2kb_AD_less_open = sum(plus2kb_overlap & direction == "AD_less_open", na.rm = TRUE),
n_plus10kb_AD_more_open = sum(plus10kb_overlap & direction == "AD_more_open", na.rm = TRUE),
n_plus10kb_AD_less_open = sum(plus10kb_overlap & direction == "AD_less_open", na.rm = TRUE),
.groups = "drop"
) %>%
arrange(desc(n_direct), desc(n_plus2kb), desc(n_plus10kb))
direct_sig <- sig %>%
filter(direct_overlap) %>%
arrange(.data[[group_col]], p_val_adj, p_val)
plus2kb_sig <- sig %>%
filter(plus2kb_overlap) %>%
arrange(.data[[group_col]], p_val_adj, p_val)
plus10kb_sig <- sig %>%
filter(plus10kb_overlap) %>%
arrange(.data[[group_col]], p_val_adj, p_val)
direct_id_summary <- direct_sig %>%
separate_rows(direct_herv_ltr_id, sep = ";") %>%
filter(!is.na(direct_herv_ltr_id), direct_herv_ltr_id != "") %>%
count(.data[[group_col]], direct_herv_ltr_id, direction, sort = TRUE)
direct_family_summary <- direct_sig %>%
separate_rows(direct_herv_ltr_id, sep = ";") %>%
filter(!is.na(direct_herv_ltr_id), direct_herv_ltr_id != "") %>%
mutate(herv_family = sub("[_-].*$", "", direct_herv_ltr_id)) %>%
count(.data[[group_col]], herv_family, direction, sort = TRUE)
wb <- createWorkbook()
addWorksheet(wb, "summary_sig")
writeData(wb, "summary_sig", summary_sig)
addWorksheet(wb, "sig_all_DARs")
writeData(wb, "sig_all_DARs", sig)
addWorksheet(wb, "sig_direct_overlap")
writeData(wb, "sig_direct_overlap", direct_sig)
addWorksheet(wb, "sig_plus2kb_overlap")
writeData(wb, "sig_plus2kb_overlap", plus2kb_sig)
addWorksheet(wb, "sig_plus10kb_overlap")
writeData(wb, "sig_plus10kb_overlap", plus10kb_sig)
addWorksheet(wb, "direct_id_summary")
writeData(wb, "direct_id_summary", direct_id_summary)
addWorksheet(wb, "direct_family_summary")
writeData(wb, "direct_family_summary", direct_family_summary)
saveWorkbook(wb, file.path(res_dir, "celltype_DAR_hERV_LTR_overlap_sig.xlsx"), overwrite = TRUE)
- 一部分显著差异开放峰直接落在hERV/LTR位点或其附近:共有153个显著DAR直接重叠hERV/LTR,376个显著DAR位于hERV/LTR±2kb范围内,1201个显著DAR位于±10kb范围内
- 这些hERV/LTR相关DAR主要分布在ASC、ODC、MG和EX中,并主要表现为AD中开放性增强
为什么用±2kb和±10kb:
- direct overlap:DAR直接落在hERV/LTR内部
- ±2kb:hERV/LTR非常近邻区域。这个DAR虽然不一定落在hERV/LTR内部,但位于hERV/LTR非常邻近的启动子/近端调控范围内
- 在这篇文章中,作者计算gene activity时也把基因体上游延伸2000bp以覆盖promoter区域
- ±10kb:局部调控环境,避免漏掉紧邻hERV/LTR但没有直接重叠的peak,例如hERV/LTR边界外几kb处有一个AD更开放的peak,可能提示该hERV/LTR所在区域整体染色质状态改变,但不能直接说“这个peak就是hERV/LTR本体开放”
- 因为这里只关注“AD中发生差异开放的ATAC peak,是否落在hERV/LTR本体或附近”,不像
2024 NC–schERV+eQTL这篇考虑所有的顺式遗传调控,所以就没有用±200kb
显著DAR是否比非显著peak更容易落在hERV/LTR区域
显著AD-DAR是否比“同一次差异分析中有机会成为显著DAR的peak”更容易落在hERV/LTR区域
- 选择的背景:同一个celltype/cluster中参与该次FindMarkers比较的所有peak
group_col <- "celltype"
padj_cutoff <- 0.05
dar <- read_xlsx(file.path(res_dir, "celltype_DAR_hERV_LTR_overlap_all.xlsx"), sheet = "all_DARs") %>%
mutate(
p_val = as.numeric(p_val),
avg_logFC = as.numeric(avg_logFC),
pct_AD = as.numeric(pct_AD),
pct_Control = as.numeric(pct_Control),
p_val_adj = as.numeric(p_val_adj),
direct_overlap = as.logical(direct_overlap),
plus2kb_overlap = as.logical(plus2kb_overlap),
plus10kb_overlap = as.logical(plus10kb_overlap),
is_sig = p_val_adj < padj_cutoff
) %>%
distinct(.data[[group_col]], peak, .keep_all = TRUE)
overlap_cols <- c("direct_overlap", "plus2kb_overlap", "plus10kb_overlap")
query_sets <- c("sig_all", "sig_AD_more_open", "sig_AD_less_open")
run_fisher <- function(df, overlap_col, query_set) {
if (query_set == "sig_all") {
query <- df$is_sig
} else if (query_set == "sig_AD_more_open") {
query <- df$is_sig & df$direction == "AD_more_open"
} else {
query <- df$is_sig & df$direction == "AD_less_open"
}
hit <- df[[overlap_col]]
a <- sum(query & hit, na.rm = TRUE)
b <- sum(query & !hit, na.rm = TRUE)
c <- sum(!query & hit, na.rm = TRUE)
d <- sum(!query & !hit, na.rm = TRUE)
mat <- matrix(c(a, b, c, d), nrow = 2, byrow = TRUE)
ft <- fisher.test(mat, alternative = "greater")
tibble(
query_set = query_set,
overlap_type = sub("_overlap$", "", overlap_col),
n_query = a + b,
n_query_overlap = a,
query_overlap_frac = ifelse(a + b > 0, a / (a + b), NA_real_),
n_background = a + b + c + d,
n_background_overlap = a + c,
background_overlap_frac = ifelse(a + b + c + d > 0, (a + c) / (a + b + c + d), NA_real_),
odds_ratio = unname(ft$estimate),
p_value = ft$p.value,
a_query_overlap = a,
b_query_not_overlap = b,
c_nonquery_overlap = c,
d_nonquery_not_overlap = d
)
}
enrichment <- dar %>%
group_by(.data[[group_col]]) %>%
group_modify(~ {
bind_rows(lapply(overlap_cols, function(oc) {
bind_rows(lapply(query_sets, function(qs) {
run_fisher(.x, oc, qs)
}))
}))
}) %>%
ungroup() %>%
rename(group = all_of(group_col)) %>%
group_by(query_set, overlap_type) %>%
mutate(FDR = p.adjust(p_value, method = "BH")) %>%
ungroup() %>%
arrange(query_set, overlap_type, FDR, p_value)
background_rate <- dar %>%
group_by(.data[[group_col]]) %>%
summarise(
n_background = n(),
n_direct_bg = sum(direct_overlap, na.rm = TRUE),
frac_direct_bg = n_direct_bg / n_background,
n_plus2kb_bg = sum(plus2kb_overlap, na.rm = TRUE),
frac_plus2kb_bg = n_plus2kb_bg / n_background,
n_plus10kb_bg = sum(plus10kb_overlap, na.rm = TRUE),
frac_plus10kb_bg = n_plus10kb_bg / n_background,
.groups = "drop"
) %>%
rename(group = all_of(group_col))
sig_rate <- dar %>%
filter(is_sig) %>%
group_by(.data[[group_col]]) %>%
summarise(
n_sig = n(),
n_direct_sig = sum(direct_overlap, na.rm = TRUE),
frac_direct_sig = n_direct_sig / n_sig,
n_plus2kb_sig = sum(plus2kb_overlap, na.rm = TRUE),
frac_plus2kb_sig = n_plus2kb_sig / n_sig,
n_plus10kb_sig = sum(plus10kb_overlap, na.rm = TRUE),
frac_plus10kb_sig = n_plus10kb_sig / n_sig,
n_sig_AD_more_open = sum(direction == "AD_more_open", na.rm = TRUE),
n_sig_AD_less_open = sum(direction == "AD_less_open", na.rm = TRUE),
.groups = "drop"
) %>%
rename(group = all_of(group_col))
sig_direct <- dar %>%
filter(is_sig, direct_overlap) %>%
arrange(.data[[group_col]], p_val_adj, p_val)
sig_plus2kb <- dar %>%
filter(is_sig, plus2kb_overlap) %>%
arrange(.data[[group_col]], p_val_adj, p_val)
sig_plus10kb <- dar %>%
filter(is_sig, plus10kb_overlap) %>%
arrange(.data[[group_col]], p_val_adj, p_val)
wb <- createWorkbook()
addWorksheet(wb, "enrichment")
writeData(wb, "enrichment", enrichment)
addWorksheet(wb, "background_rate")
writeData(wb, "background_rate", background_rate)
addWorksheet(wb, "sig_rate")
writeData(wb, "sig_rate", sig_rate)
addWorksheet(wb, "sig_direct_overlap")
writeData(wb, "sig_direct_overlap", sig_direct)
addWorksheet(wb, "sig_plus2kb_overlap")
writeData(wb, "sig_plus2kb_overlap", sig_plus2kb)
addWorksheet(wb, "sig_plus10kb_overlap")
writeData(wb, "sig_plus10kb_overlap", sig_plus10kb)
saveWorkbook(wb, file.path(res_dir, "celltype_DAR_hERV_LTR_enrichment.xlsx"), overwrite = TRUE)
- odds_ratio > 1:显著DAR中hERV/LTR重叠比例高于背景
- FDR < 0.05:在多重检验校正后,富集比较可靠
- query_overlap_frac > background_overlap_frac:方向上支持富集
- 在celltype层面,显著AD-DAR与hERV/LTR区域的重叠比例与背景peak接近,未观察到显著富集;但MG和ASC中AD开放增强DAR在hERV/LTR直接重叠或近邻区域存在轻微富集趋势
- 与hERV/LTR直接重叠的显著DAR,大多数是AD中开放增强,尤其MG、ODC、EX全部是AD_more_open
联合分析hERV的DE和DAR表达结果
我用之前的方法单独做了这个数据集的位点级差异表达,与DAR结果对齐:预览 | 下载
herv_padj_cutoff <- 0.05
# 读取ATAC结果
atac_sig <- read_xlsx(file.path(res_dir, "celltype_DAR_hERV_LTR_overlap_sig.xlsx"), sheet = "sig_all_DARs") %>%
mutate(
p_val = as.numeric(p_val),
avg_logFC = as.numeric(avg_logFC),
p_val_adj = as.numeric(p_val_adj),
direct_overlap = as.logical(direct_overlap),
plus2kb_overlap = as.logical(plus2kb_overlap),
plus10kb_overlap = as.logical(plus10kb_overlap)
)
make_overlap_long <- function(df, overlap_type, flag_col, id_col) {
df %>%
filter(.data[[flag_col]]) %>%
mutate(
overlap_type = overlap_type,
herv_ltr_id = .data[[id_col]]
) %>%
filter(!is.na(herv_ltr_id), herv_ltr_id != "") %>%
separate_rows(herv_ltr_id, sep = ";")
}
atac_long <- bind_rows(
make_overlap_long(atac_sig, "direct", "direct_overlap", "direct_herv_ltr_id"),
make_overlap_long(atac_sig, "plus2kb", "plus2kb_overlap", "plus2kb_herv_ltr_id"),
make_overlap_long(atac_sig, "plus10kb", "plus10kb_overlap", "plus10kb_herv_ltr_id")
) %>%
rename(
atac_celltype = celltype,
atac_p_val = p_val,
atac_logFC = avg_logFC,
atac_p_val_adj = p_val_adj,
atac_direction = direction
)
# 读取hERV表达DE结果并统一细胞类型名称
herv_de <- read.csv("C:\\Users\\17185\\Desktop\\file\\ATACtest\\data\\hERV_locus_celltype.csv", check.names = FALSE) %>%
filter(celltype != "All") %>%
mutate(
p_val = as.numeric(p_val),
avg_log2FC = as.numeric(avg_log2FC),
p_val_adj = as.numeric(p_val_adj),
atac_celltype = recode(
celltype,
"Astro" = "ASC",
"Excit" = "EX",
"Inhib" = "INH",
"Mic" = "MG",
"Oligo" = "ODC",
"OPC" = "OPC",
"Endo" = "PER.END"
),
herv_expr_direction = case_when(
avg_log2FC > 0 ~ "AD_up",
avg_log2FC < 0 ~ "AD_down",
TRUE ~ "no_change"
),
herv_expr_sig = p_val_adj < herv_padj_cutoff
) %>%
rename(
herv_locus = feature,
herv_expr_p_val = p_val,
herv_expr_log2FC = avg_log2FC,
herv_expr_p_val_adj = p_val_adj,
herv_pct_AD = pct.1,
herv_pct_NC = pct.2
)
herv_de$herv_locus <- gsub("-", "_", herv_de$herv_locus)
# 取交集
intersect_all <- inner_join(
atac_long,
herv_de,
by = c("atac_celltype", "herv_ltr_id" = "herv_locus")
) %>%
mutate(
evidence_level = case_when(
overlap_type == "direct" ~ "DAR_inside_hERV_LTR",
overlap_type == "plus2kb" ~ "DAR_near_hERV_LTR_2kb",
overlap_type == "plus10kb" ~ "DAR_near_hERV_LTR_10kb"
),
direction_relation = case_when(
atac_direction == "AD_more_open" & herv_expr_direction == "AD_up" ~ "open_up_expr_up",
atac_direction == "AD_less_open" & herv_expr_direction == "AD_down" ~ "open_down_expr_down",
atac_direction == "AD_more_open" & herv_expr_direction == "AD_down" ~ "open_up_expr_down",
atac_direction == "AD_less_open" & herv_expr_direction == "AD_up" ~ "open_down_expr_up",
TRUE ~ "other"
)
) %>%
arrange(atac_celltype, overlap_type, herv_expr_p_val_adj, atac_p_val_adj) %>%
rename(herv_locus = herv_ltr_id)
# 筛选表达也显著的交集
intersect_expr_sig <- intersect_all %>%
filter(herv_expr_sig) %>%
arrange(atac_celltype, overlap_type, herv_expr_p_val_adj, atac_p_val_adj)
# 因为direct也会被包含在plus2kb/plus10kb里,为了避免重复,可以做一个“最佳证据等级”表:同一个celltype-peak-hERV只保留最严格的overlap类型
intersect_best <- intersect_expr_sig %>%
mutate(overlap_rank = case_when(
overlap_type == "direct" ~ 1,
overlap_type == "plus2kb" ~ 2,
overlap_type == "plus10kb" ~ 3
)) %>%
group_by(atac_celltype, peak, herv_locus) %>%
arrange(overlap_rank, herv_expr_p_val_adj, atac_p_val_adj, .by_group = TRUE) %>%
slice_head(n = 1) %>%
ungroup() %>%
select(-overlap_rank)
# 汇总结果
summary_by_celltype <- intersect_best %>%
group_by(atac_celltype, overlap_type, direction_relation) %>%
summarise(
n_pairs = n(),
n_hERV_loci = n_distinct(herv_locus),
n_DAR_peaks = n_distinct(peak),
.groups = "drop"
) %>%
arrange(atac_celltype, overlap_type, desc(n_pairs))
summary_by_herv <- intersect_best %>%
group_by(herv_locus) %>%
summarise(
n_celltypes = n_distinct(atac_celltype),
celltypes = paste(sort(unique(atac_celltype)), collapse = ";"),
overlap_types = paste(sort(unique(overlap_type)), collapse = ";"),
direction_relations = paste(sort(unique(direction_relation)), collapse = ";"),
min_herv_expr_padj = min(herv_expr_p_val_adj, na.rm = TRUE),
min_atac_padj = min(atac_p_val_adj, na.rm = TRUE),
.groups = "drop"
) %>%
arrange(min_herv_expr_padj, min_atac_padj)
priority_candidates <- intersect_best %>%
filter(
overlap_type %in% c("direct", "plus2kb"),
direction_relation %in% c("open_up_expr_up", "open_down_expr_down")
) %>%
arrange(overlap_type, atac_celltype, herv_expr_p_val_adj, atac_p_val_adj)
wb <- createWorkbook()
addWorksheet(wb, "summary_by_celltype")
writeData(wb, "summary_by_celltype", summary_by_celltype)
addWorksheet(wb, "summary_by_hERV")
writeData(wb, "summary_by_hERV", summary_by_herv)
addWorksheet(wb, "all_intersections")
writeData(wb, "all_intersections", intersect_all)
addWorksheet(wb, "expr_sig_intersections")
writeData(wb, "expr_sig_intersections", intersect_expr_sig)
addWorksheet(wb, "best_intersections")
writeData(wb, "best_intersections", intersect_best)
addWorksheet(wb, "priority_candidates")
writeData(wb, "priority_candidates", priority_candidates)
saveWorkbook(wb, file.path(res_dir, "celltype_hERV_expr_ATAC_intersect.xlsx"), overwrite = TRUE)
- priority_candidates:最核心。这里面的位点同时有ATAC证据和表达证据,而且方向一致
- summary_by_celltype:看哪类细胞中交集最多,比如MG、ODC、ASC是否更突出
- summary_by_celltype:看哪类细胞中交集最多,比如MG、ODC、ASC是否更突出
- best_intersections:完整交集表
- ATAC显著DAR与hERV/LTR位点重叠,且能在hERV表达DE表中找到对应位点:86个
- 上述交集中,hERV表达也显著差异:10个
- 去掉direct/±2kb/±10kb重复:6个
- direct/±2kb且ATAC和表达方向一致:0个
- 综上所述,细胞类型层面存在“ATAC显著DAR+hERV表达显著DE”的交集,但严格的“近端/本体开放变化与表达变化方向一致”候选暂时没有;方向一致的候选目前都在±10kb范围内
| celltype | overlap类型 | hERV位点 | ATAC方向 | hERV表达方向 | 关系 |
|---|---|---|---|---|---|
| MG(mic) | ±10kb | HARLEQUIN_4q32.1 | AD更开放 | AD下调 | 不一致 |
| MG | ±10kb | HARLEQUIN_4q32.1 | AD更开放 | AD下调 | 不一致 |
| ODC(oligo) | ±10kb | MER41_14q32.13 | AD更开放 | AD上调 | 一致 |
| ODC | direct | MER41_16q21 | AD更开放 | AD下调 | 不一致 |
| ODC | direct | ERV316A3_21q22.3e | AD更开放 | AD下调 | 不一致 |
| ODC | ±10kb | ERVLE_2q35a | AD更开放 | AD上调 | 一致 |
不能简单用“开放性增强→表达上调”解释:
- 这个ATAC peak反映的是局部调控元件开放,而不是hERV自身转录启动
- 该位点可能位于hERV内部某个片段,开放性变化与完整hERV转录方向不一致
- hERV表达受其它调控层影响,例如DNA甲基化、H3K9me3等
- 该hERV可能嵌在某个宿主基因或调控区域中,ATAC信号更多反映宿主区域状态
注释附近基因
# 读取6个候选位点
cand <- read_xlsx(file.path(res_dir, "celltype_hERV_expr_ATAC_intersect.xlsx"), sheet = "best_intersections") %>%
mutate(
candidate_id = paste(atac_celltype, herv_locus, peak, overlap_type, sep = "__"),
candidate_id = str_replace_all(candidate_id, "[^A-Za-z0-9_.:-]+", "_")
) %>%
distinct(candidate_id, .keep_all = TRUE)
norm_id <- function(x) {
x %>%
as.character() %>%
str_replace_all("[^A-Za-z0-9]", "") %>%
toupper()
}
# 读取hERV BED并匹配候选位点坐标
herv_gr_all <- import("C:\\Users\\17185\\Desktop\\file\\ATACtest\\data\\hERV_locus.bed")
seqlevelsStyle(herv_gr_all) <- "UCSC"
herv_name <- if ("name" %in% colnames(mcols(herv_gr_all))) {
mcols(herv_gr_all)$name
} else if (!is.null(names(herv_gr_all))) {
names(herv_gr_all)
} else {
paste0(seqnames(herv_gr_all), ":", start(herv_gr_all), "-", end(herv_gr_all))
}
mcols(herv_gr_all)$herv_locus <- herv_name
mcols(herv_gr_all)$herv_locus_norm <- norm_id(herv_name)
cand <- cand %>%
mutate(herv_locus_norm = norm_id(herv_locus))
herv_match <- data.frame(
herv_locus = mcols(herv_gr_all)$herv_locus,
herv_locus_norm = mcols(herv_gr_all)$herv_locus_norm,
herv_chr = as.character(seqnames(herv_gr_all)),
herv_start = start(herv_gr_all),
herv_end = end(herv_gr_all),
herv_strand = as.character(strand(herv_gr_all))
)
cand2 <- cand %>%
left_join(herv_match, by = "herv_locus_norm", suffix = c("", "_bed"))
# 把DAR peak转成坐标
peak_to_df <- function(peaks) {
peaks <- as.character(peaks)
x <- if (any(grepl(":", peaks))) {
str_match(peaks, "^(.+):(\\d+)-(\\d+)$")
} else {
str_match(peaks, "^(.+)-(\\d+)-(\\d+)$")
}
tibble(
peak = peaks,
peak_chr = x[, 2],
peak_start = as.integer(x[, 3]),
peak_end = as.integer(x[, 4])
)
}
peak_df <- peak_to_df(cand2$peak)
cand2 <- cand2 %>%
select(-any_of(c("peak_chr", "peak_start", "peak_end"))) %>%
left_join(peak_df, by = "peak") %>%
mutate(
peak_mid = floor((peak_start + peak_end) / 2),
herv_mid = floor((herv_start + herv_end) / 2),
peak_to_hERV_distance = case_when(
peak_chr != herv_chr ~ NA_integer_,
peak_end < herv_start ~ herv_start - peak_end,
peak_start > herv_end ~ peak_start - herv_end,
TRUE ~ 0L
)
)
# 读取GENCODE gene注释
gtf_gene <- fread("C:\\Users\\17185\\Desktop\\file\\ATACtest\\data\\gencode.v49.annotation.gtf.gz", sep = "\t", header = FALSE, quote = "", data.table = FALSE)
colnames(gtf_gene) <- c(
"seqname", "source", "feature", "start", "end",
"score", "strand", "frame", "attribute"
)
get_attr <- function(x, key) {
str_match(x, paste0(key, ' "([^"]+)"'))[, 2]
}
gene_df <- gtf_gene %>%
transmute(
chr = seqname,
start = as.integer(start),
end = as.integer(end),
strand = strand,
gene_id = get_attr(attribute, "gene_id"),
gene_name = get_attr(attribute, "gene_name"),
gene_type = coalesce(
get_attr(attribute, "gene_type"),
get_attr(attribute, "gene_biotype")
)
) %>%
filter(!is.na(gene_name))
gene_gr <- GRanges(
seqnames = gene_df$chr,
ranges = IRanges(gene_df$start, gene_df$end),
strand = gene_df$strand
)
mcols(gene_gr)$gene_id <- gene_df$gene_id
mcols(gene_gr)$gene_name <- gene_df$gene_name
mcols(gene_gr)$gene_type <- gene_df$gene_type
# 构建hERV和peak的GRanges
herv_gr <- GRanges(
seqnames = cand2$herv_chr,
ranges = IRanges(cand2$herv_start, cand2$herv_end),
strand = cand2$herv_strand
)
mcols(herv_gr)$candidate_id <- cand2$candidate_id
mcols(herv_gr)$herv_locus <- cand2$herv_locus
mcols(herv_gr)$atac_celltype <- cand2$atac_celltype
peak_gr <- GRanges(
seqnames = cand2$peak_chr,
ranges = IRanges(cand2$peak_start, cand2$peak_end)
)
mcols(peak_gr)$candidate_id <- cand2$candidate_id
mcols(peak_gr)$peak <- cand2$peak
mcols(peak_gr)$herv_locus <- cand2$herv_locus
mcols(peak_gr)$atac_celltype <- cand2$atac_celltype
# 最近基因和±50kb基因注释
nearest_gene <- function(query_gr, query_type) {
hit <- distanceToNearest(query_gr, gene_gr, ignore.strand = TRUE)
tibble(
candidate_id = mcols(query_gr)$candidate_id[queryHits(hit)],
query_type = query_type,
query_chr = as.character(seqnames(query_gr))[queryHits(hit)],
query_start = start(query_gr)[queryHits(hit)],
query_end = end(query_gr)[queryHits(hit)],
gene_id = mcols(gene_gr)$gene_id[subjectHits(hit)],
gene_name = mcols(gene_gr)$gene_name[subjectHits(hit)],
gene_type = mcols(gene_gr)$gene_type[subjectHits(hit)],
gene_chr = as.character(seqnames(gene_gr))[subjectHits(hit)],
gene_start = start(gene_gr)[subjectHits(hit)],
gene_end = end(gene_gr)[subjectHits(hit)],
gene_strand = as.character(strand(gene_gr))[subjectHits(hit)],
distance = mcols(hit)$distance
) %>%
left_join(cand2 %>% select(candidate_id, atac_celltype, herv_locus, peak, overlap_type, atac_direction, herv_expr_direction, direction_relation), by = "candidate_id")
}
genes_in_flank <- function(query_gr, query_type, flank = 50000) {
win_gr <- query_gr
start(win_gr) <- pmax(start(win_gr) - flank, 1)
end(win_gr) <- end(win_gr) + flank
ov <- findOverlaps(win_gr, gene_gr, ignore.strand = TRUE)
tibble(
candidate_id = mcols(query_gr)$candidate_id[queryHits(ov)],
query_type = query_type,
flank_bp = flank,
query_chr = as.character(seqnames(query_gr))[queryHits(ov)],
query_start = start(query_gr)[queryHits(ov)],
query_end = end(query_gr)[queryHits(ov)],
gene_id = mcols(gene_gr)$gene_id[subjectHits(ov)],
gene_name = mcols(gene_gr)$gene_name[subjectHits(ov)],
gene_type = mcols(gene_gr)$gene_type[subjectHits(ov)],
gene_chr = as.character(seqnames(gene_gr))[subjectHits(ov)],
gene_start = start(gene_gr)[subjectHits(ov)],
gene_end = end(gene_gr)[subjectHits(ov)],
gene_strand = as.character(strand(gene_gr))[subjectHits(ov)],
distance = distance(query_gr[queryHits(ov)], gene_gr[subjectHits(ov)])
) %>%
left_join(cand2 %>% select(candidate_id, atac_celltype, herv_locus, peak, overlap_type, atac_direction, herv_expr_direction, direction_relation), by = "candidate_id") %>%
arrange(candidate_id, distance, gene_name)
}
nearest_herv <- nearest_gene(herv_gr, "hERV_locus")
genes50_herv <- genes_in_flank(herv_gr, "hERV_locus", flank = 50000)
nearest_peak <- nearest_gene(peak_gr, "DAR_peak")
genes50_peak <- genes_in_flank(peak_gr, "DAR_peak", flank = 50000)
# 汇总每个候选位点的最近基因和±50kb基因
collapse_gene <- function(df) {
df %>%
group_by(candidate_id) %>%
summarise(
genes_50kb = paste(unique(gene_name), collapse = ";"),
n_genes_50kb = n_distinct(gene_name),
.groups = "drop"
)
}
summary_table <- cand2 %>%
left_join(
nearest_herv %>%
select(candidate_id, nearest_gene_hERV = gene_name,
nearest_gene_hERV_type = gene_type,
nearest_gene_hERV_distance = distance),
by = "candidate_id"
) %>%
left_join(
collapse_gene(genes50_herv) %>%
dplyr::rename(genes_50kb_hERV = genes_50kb,
n_genes_50kb_hERV = n_genes_50kb),
by = "candidate_id"
) %>%
left_join(
nearest_peak %>%
select(candidate_id, nearest_gene_peak = gene_name,
nearest_gene_peak_type = gene_type,
nearest_gene_peak_distance = distance),
by = "candidate_id"
) %>%
left_join(
collapse_gene(genes50_peak) %>%
dplyr::rename(genes_50kb_peak = genes_50kb,
n_genes_50kb_peak = n_genes_50kb),
by = "candidate_id"
) %>%
arrange(atac_celltype, overlap_type, herv_locus)
wb <- createWorkbook()
addWorksheet(wb, "candidate_summary")
writeData(wb, "candidate_summary", summary_table)
addWorksheet(wb, "nearest_gene_hERV")
writeData(wb, "nearest_gene_hERV", nearest_herv)
addWorksheet(wb, "genes_50kb_hERV")
writeData(wb, "genes_50kb_hERV", genes50_herv)
addWorksheet(wb, "nearest_gene_peak")
writeData(wb, "nearest_gene_peak", nearest_peak)
addWorksheet(wb, "genes_50kb_peak")
writeData(wb, "genes_50kb_peak", genes50_peak)
saveWorkbook(
wb,
file.path(res_dir, "candidate_gene_annotation.xlsx"),
overwrite = TRUE
)
| hERV位点 | 细胞类型 | overlap | ATAC方向 | hERV表达方向 | 最近/重叠gene | 关系 |
|---|---|---|---|---|---|---|
| HARLEQUIN_4q32.1 | MG | ±10kb,两个peak | AD更开放 | AD下调 | TMEM144 | 方向不一致 |
| ERV316A3_21q22.3e | ODC | direct | AD更开放 | AD下调 | ENSG00000289421,附近DIP2A/PCNT | 方向不一致 |
| MER41_16q21 | ODC | direct | AD更开放 | AD下调 | CDH11 | 方向不一致 |
| ERVLE_2q35a | ODC | ±10kb | AD更开放 | AD上调 | RPL37A,附近SMARCAL1 | 方向一致 |
| MER41_14q32.13 | ODC | ±10kb | AD更开放 | AD上调 | CLMN | 方向一致 |
- MER41_14q32.13,ODC,CLMN区域:AD中附近peak开放增强,hERV表达上调,且hERV与CLMN反向
- ERVLE_2q35a,ODC,RPL37A/SMARCAL1区域:距离peak很近,表达上调非常显著,但因为它和RPL37A同向,需要排查是否受宿主gene转录影响
- ERV316A3_21q22.3e,ODC,ENSG00000289421/DIP2A/PCNT区域:direct overlap,ATAC上调,但表达下调
- MER41_16q21,ODC,CDH11区域:direct overlap,但ATAC效应量较小,DE和DAR结果反向
- HARLEQUIN_4q32.1,MG,TMEM144区域:有两个附近DAR,说明局部开放改变比较稳定,但表达下调
hERV区域开放性与转录变化并不总是同向:很可能短/不稳定herv表达是上调的,受开放性及去甲基化影响的;但是真正能形成稳定lncRNA的herv,可能比较少,而且或许其受到的调控不是这么简单的“开放性增强”
- 或许可以适当放宽DAR的标准,有更多的DAR,再试一次,确认结论不是因为统计数据问题
- 对比两个数据集分别的位点DE结果,看一致性如何,有没有一些偏稳定的信号
差异表达的hERV:
- 除了开放性,是否其5’LTR及附近区域受某些AD中变化的TF调控,这是较大可能性了
- 也可能就是噪音,为了确定不是噪音,需要看看其他AD数据集能不能都找到类似的herv
- 是不是别人在bulk rna-seq里找到的那些HERV会与DAR更一致
retry
首先看一下两个数据集分别的位点DE结果,以下表格展示了GSE157827/GSE174367/合并~MAST/LR的分析结果
| 细胞类型 | hERV位点 | 方向 | GSE157827 log2FC | GSE174367 log2FC | 解读 |
|---|---|---|---|---|---|
| Excit | HARLEQUIN-1p34.3b | 上调 | 3.46 | 1.34 | 神经元中较稳定,上调幅度大 |
| Inhib | HARLEQUIN-1p34.3b | 上调 | 3.10 | 0.76 | 抑制性神经元中也重复出现 |
| Oligo | HERV3-1p34.3 | 上调 | 4.96 | 0.77 | 少突胶质细胞中稳定,但GSE174367效应较弱 |
| Oligo | MER61-21q21.1a | 下调 | -0.35 | -0.29 | 少突胶质细胞中稳定下调,两个数据集方向一致 |
| Oligo | MER61-21q21.1b | 下调 | -0.36 | -0.29 | 与上一个位点构成21q21.1下调小簇 |
| Oligo | MER41-16q21 | 下调 | -0.30 | -0.40 | 少突胶质细胞稳定下调 |
| Astro | HML3-1p32.3 | 下调 | -0.33 | -0.42 | 星形胶质细胞中较稳定,下调方向一致 |
其它一些比较有争议的:
| 位点 | 问题 |
|---|---|
| HML3-1q32.1b | 在GSE157827多个细胞类型下调,但在GSE174367的Excit/All反而上调 |
| HML1-1q32.1 | 和HML3-1q32.1b类似,两个数据集方向冲突 |
| HML4-16p13.3 | 方向大体偏下调,但效应较小,LR下不够稳定 |
| HERVW-15q21.2 | 主要由GSE174367和合并结果支持,GSE157827中不够稳定 |
| HARLEQUIN-6p21.31 | 合并结果看起来有信号,但分数据集/分方法后不够稳定,尤其LR支持弱 |
综合了基因组位置类型注释后:
| 细胞类型 | hERV位点 | 表达方向 | 位置/邻近基因 | 初步解释 |
|---|---|---|---|---|
| Excit/Inhib/All | HARLEQUIN-1p34.3b |
AD上调 | intergenic,近SFPQ约43.6kb |
跨神经元重复出现,像稳定表达标志物 |
| All/Oligo | HERV3-1p34.3 |
AD上调 | intergenic,近SFPQ约46.9kb |
和上一个构成1p34.3区域候选 |
| Astro | HML3-1p32.3 |
AD下调 | exonic_partial_same,ACOT11
|
更像gene-hERV嵌合/isoform候选 |
| Oligo | MER41-16q21 |
AD下调 | intronic_full_same,CDH11
|
有局部DAR支持,但方向相反 |
| Oligo | MER61-21q21.1a/b |
AD下调 | intergenic,近NCAM2约217–220kb |
Oligo稳定下调候选,但没有近端DAR支持 |
- HERV3-1p34.3和HARLEQUIN-1p34.3b都在All层面稳定上调,而且HARLEQUIN-1p34.3b还在Excit和Inhib中稳定上调;两者都位于SFPQ附近的基因间区,但与SFPQ的表达相关性很低,不像是简单的SFPQ转录外溢。但在DAR层面,这两个位点在对应细胞类型中没有显著DAR直接重叠:更像稳定转录变化信号,而不是局部染色质开放增强直接解释的信号
- MER41-16q21在Oligo中两个数据集都稳定下调,是CDH11同方向内含子(和CDH11表达相关性较高),在hERV内部重叠显著DAR,但DAR为AD_more_open
//todo 画韦恩图看不同数据集的差异是否一致 看一下邻近基因的功能? 差异表达hERV找一下bulk的
后续思路
Systematic characterization of the disease-relevant regulation of putatively full-length HERV transcripts in Alzheimer’s cortex: (1) 差异表达定总集,细胞特异性刻画;(2)定出潜在类别(如嵌合转录本型、regional表观驱动型、复杂调控型);(3)刻画各类别功能(共表达);(4)挖掘各类别上游调控因素,如TF, SNP, 与AD疾病挂钩解释相关性。
找AD疾病关联/因果HERV,在我们缺少配套genotype的情况下,或许可以这样做:(1)HERV~SNP的QTL(cortex,别人已经做好的),多个AD的GWAS summary(AD~SNP),做SMR,鉴定SNP~HERV~AD关系;(2)你算出来的AD cortex中HERV ~ gene关系,加上gene~SNP的cortex的eQTL,两个都是回归模型,理论上可以算出AD状态下HERV~SNP的关系;(3)整合(1)(2)