暑假前
**— layout: mypost title: 暑假前文献阅读和计划安排 category: other subcategory: other-other —
暑假前文献阅读和计划安排
文献阅读
Integrated multimodal cell atlas of Alzheimer’s disease
- 全球首个阿尔茨海默病多模态细胞图谱
- 如果HERV在一个基因内部(同义链),且基因本身也表达,则有可能是构成了这个基因转录本的一个Isoform; 如果该基因表达低、HERV在反义链、或HERV周围没有基因,则更可能是它自己的转录
- 这些microglia-like cells (小胶质样脑巨噬细胞)跟你在mic里看到的亚组有关嘛
stellarscope原文:A single-cell transposable element atlas of human cell identity
- 用hERV定义细胞类型
- 分析非常全面 可以效仿 就是生物学上差点意思
2025 NG–A retrotransposon caught red-handed in a curious case of missing digits
- 这篇论文提到了一个比较有趣的方法,用现有的单细胞大模型Geneformer打底,用特定的case/control数据做微调,然后做in silico perturbation找关键基因。可学习下这个思路
Classification and characterization of human endogenous retroviruses; mosaic forms are common
- 近期重磅论文比如cell等里引用参考的HERV的分类
包含脑在内的单细胞atac:https://health.tsinghua.edu.cn/human-scatac-corpus/download.php
2024 NC–hERV + TWAS
样本情况:CommonMind Consortium(CMC)数据集,人类死后脑的背外侧前额叶皮层(DLPFC)
- 初始样本:910个,表达数据、基因型数据、临床信息,包含无精神疾病诊断442人、精神分裂症350人、双相情感障碍110人
- 基因型数据:SNP基因型矩阵,比如每个人在某个位点是AA、AG还是GG
- 选择了两个较大的、相对同质的祖源群体:欧洲血统共563人+非洲血统共229人,都是包含患病和健康
- “把死亡时有精神疾病诊断的个体也纳入SNP权重构建,是因为增加样本量能提高检测cis调控效应的能力”
- “结果显示,使用完整欧洲样本比只用242个未受影响个体更有统计功效,且两者rTWAS结果高度一致”
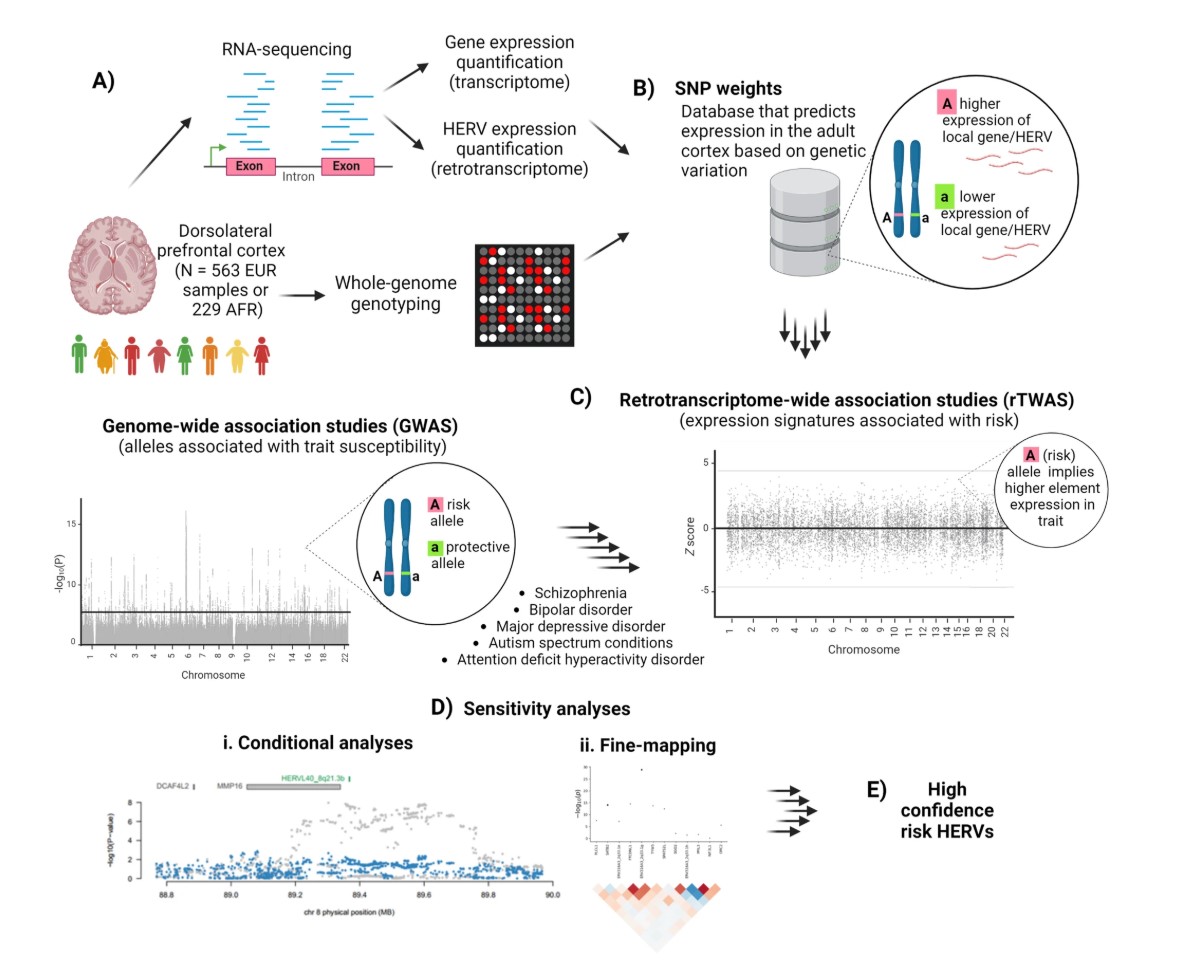
数据预处理:
- 基因型QC:去除了杂合率异常的样本、亲缘关系较高的样本、基因型缺失率较高的样本,以及遗传性别和记录性别不一致的样本
- 杂合率异常:可能提示样本污染、测序/芯片质量问题或群体结构异常
- pihat>0.2:PLINK中估计的样本亲缘关系指标,>0.2说明两个人可能有较近亲缘关系。TWAS建模一般希望样本近似独立
- fastq处理:Trimmomatic去除低质量碱基和过短reads
第1步:在DLPFC脑组织中定量gene和HERV表达
- 位点级HERV表达定量:Bowtie2 + Telescope
- 常规基因表达定量:
- kallisto不做传统逐碱基全比对,而是pseudoalignment,速度快,适合转录本/基因表达定量
- lengthScaledTPM是tximport中常用设置,用TPM回推count,同时考虑长度校正,方便后续统计模型使用
- biomaRt用于把Ensembl等数据库注释转换成标准gene信息
- 作者把有HUGO Gene Nomenclature Committee gene symbol的基因定义为canonical genes(常规基因/标准基因),主要是蛋白编码基因
- 表达过滤:把HERV和gene表达矩阵合并,然后只保留在至少20%样本中满足read counts≥6且TPM≥0.1的feature
- 这个阈值来自GTEx eQTL分析习惯,目的是去掉极低表达、难以可靠建模的feature。对于TWAS来说,表达太稀疏的feature无法稳定估计SNP weights
- 校正协变量:用limma调整表达数据,协变量包括样本来源机构、病例/对照状态、RIN、性别、死后间隔、年龄分组、前10个祖源PC,以及sva计算的surrogate variables
- limma:常用于表达矩阵线性模型校正
- RNA完整性指标(RIN):死后脑RNA质量差异很大,必须校正
- 死亡到取材间隔(post-mortem interval, PMI):会影响RNA降解
- 祖源主成分(population PCs):用于控制群体结构
- 未知批次效应或隐含混杂因素(surrogate variables, sva):例如实验批次、RNA质量残留差异、未记录的技术因素等
- 年龄分箱:作者把年龄分为17–29、30–49、50–69、70–89、90+,而不是直接使用连续年龄,可能是为了处理非线性年龄效应和>90岁这种顶码记录
第2步:构建SNP weights(遗传变异预测表达)模型
- Fig.1B:在某个局部基因组区域中,如果某个SNP的A等位基因对应某个gene/HERV更高表达,那么就可以学习出这个SNP对表达的权重。“用附近SNP预测某个gene/HERV表达量”
- FUSION TWAS权重计算:在欧洲/非洲CMC子集中分别估计每个gene/HERV的cis遗传调控模型,Expressiongene/HERV ≈ β1SNP1 + β2SNP2 + … + βkSNPk,其中这些SNP都在该feature附近1Mb内
- 其中使用了BLUP/BSLMM/LASSO/Elastic net/Top SNP模型,这些模型对应不同遗传架构假设。某个HERV表达可能由一个强SNP控制,也可能由多个弱SNP共同控制。FUSION会尝试多种模型,从而提高不同类型feature的预测能力
- eQTL分析通常逐个SNP-feature配对检验,而TWAS权重构建更像是构建一个由多个cis SNP共同预测表达的模型
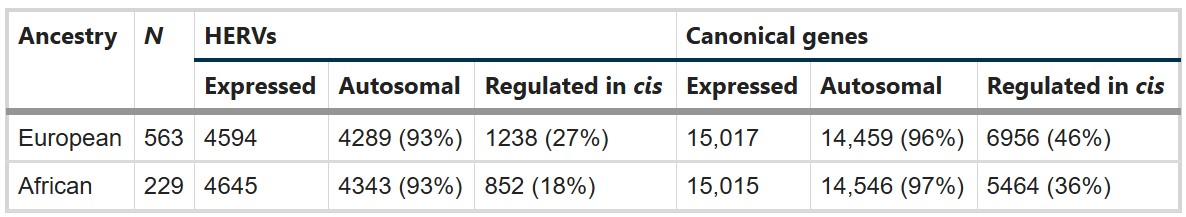
- 在DLPFC中能检测到多少HERV?其中多少有cis遗传调控?
- 欧洲血统563个样本中,检测到4594个表达HERV,其中4289个位于常染色体,1238个有显著cis遗传调控;有15017个表达基因,6956个有cis调控
- 非洲血统229个样本中,检测到4645个表达HERV,其中4343个位于常染色体,852个有显著cis遗传调控;有15015个表达基因,5464个有cis调控
- 成人DLPFC中确实存在大量可检测HERV表达,而且相当一部分HERV表达能被附近遗传变异调控,为后面做rTWAS提供了前提
- 非洲组表达HERV数量和欧洲组差不多,但cis-heritable HERV数量更少,作者认为这可能部分来自样本量差异,也可能提示祖源差异
第3步:把SNP weights和精神疾病GWAS结果整合,做rTWAS
- Fig.1C:把表达预测模型和精神疾病GWAS summary statistics交叉整合,找出“被遗传风险牵引的表达特征”
- TWAS:把表达预测模型和GWAS summary statistics整合
- rTWAS:把TWAS对象从普通gene扩展到HERV
- 输入:
- 每个gene/HERV的SNP weights
- 精神疾病GWAS summary statistics:只包含每个SNP的效应值、标准误、P值等,不需要个体级数据
- LD reference panel,即与建权重样本相匹配的CMC基因型数据:如果不控制LD,一个区域内多个SNP信号会重复计入
-
FUSION:用GWAS SNP效应和表达预测权重推断“遗传上预测的表达”是否与疾病风险相关
如果某个HERV附近的SNP既能预测HERV表达,又在GWAS中影响疾病风险,那么该HERV就可能出现rTWAS显著
- TWAS Z-score方向:正值通常表示遗传风险相关等位基因预测更高表达,负值表示预测更低表达,不等同于病例脑组织真实表达升降
- Bonferroni校正:严格多重检验校正,用于控制全局假阳性
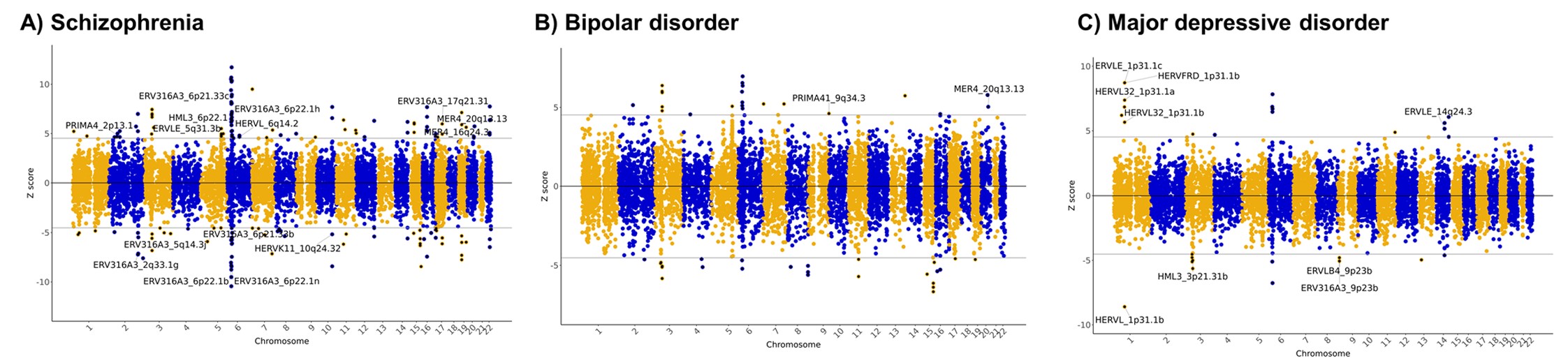
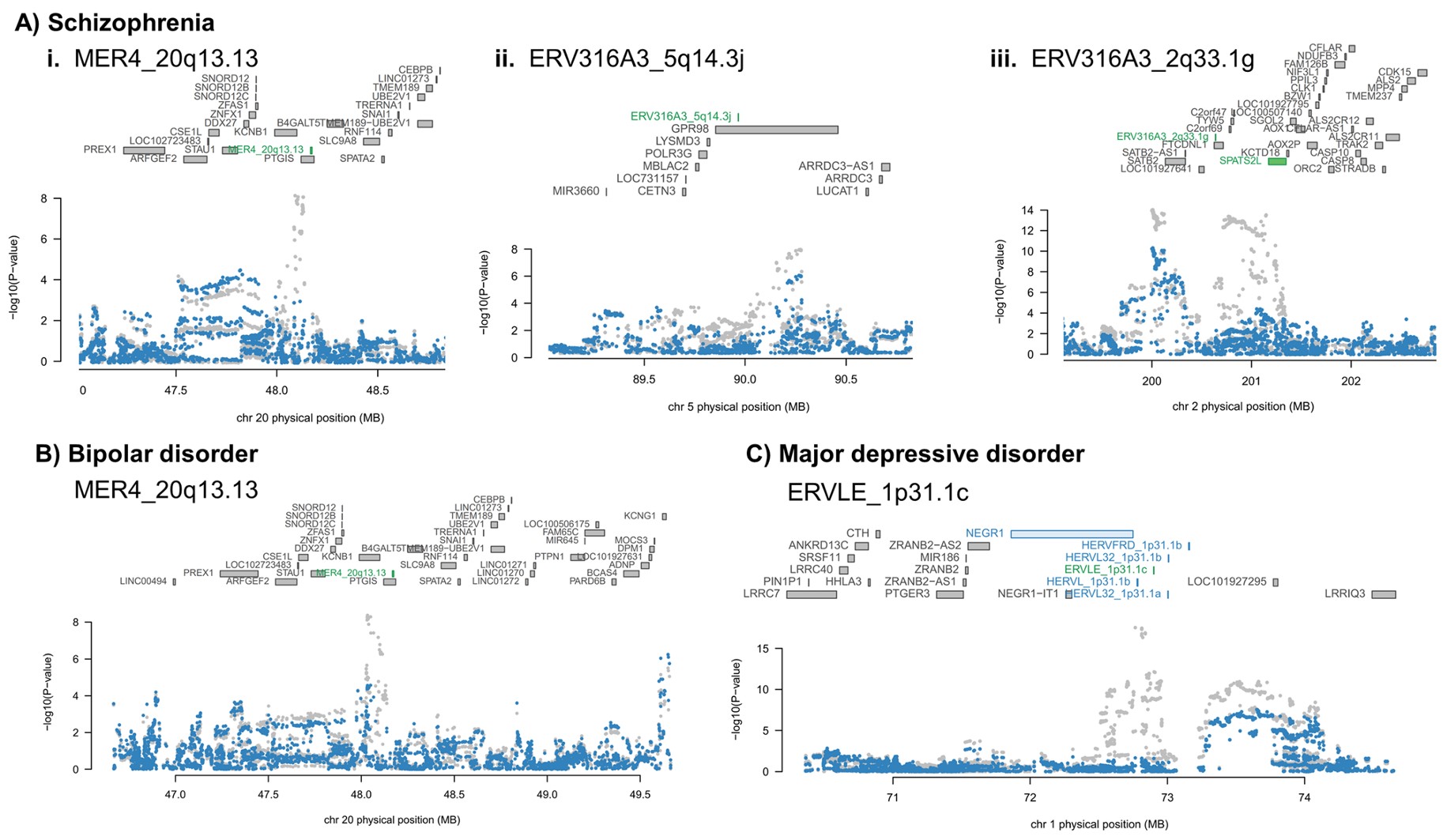
- Manhattan plot(曼哈顿图):分别对应精神分裂症、双相情感障碍和重度抑郁障碍。横轴是基因组位置,纵轴是TWAS Z-score;灰色水平线是Bonferroni显著性阈值。这里找的特征同时包括基因和hERV,图上标注的是其中的hERV位点部分
- MER4_20q13.13也在双相情感障碍和精神分裂症rTWAS中出现,且方向一致,提示MER4_20q13.13可能对应精神分裂症和双相障碍的共同遗传风险机制
- 抑郁症中HERV占比看起来较高,但后续真正进入高可信名单的主要是ERVLE_1p31.1c
- ADHD和自闭症谱系障碍没有显著HERV信号
- 这里的“风险表达特征”不是说病例中真实表达一定升高/降低,而是说遗传上预测的表达水平与疾病GWAS风险相关
- 这里出现的HERV信号需要后续条件分析判断它是不是独立于周围基因
第4步:用条件分析和fine-mapping筛出更可信的HERV
- Fig.1D/E:先看某个HERV信号是否独立于邻近gene,再看它在同一LD区域中是否最可能解释GWAS信号。最后得到high confidence risk HERVs
- 连锁不平衡(linkage disequilibrium, LD):附近SNP常一起遗传,所以一个GWAS峰可能牵连多个gene/HERV表达信号
- 后验纳入概率(posterior inclusion probability, PIP):PIP>0.5表示该表达特征比同一区域其它特征更可能解释关联信号
- 条件分析(conditional analyses):把同一区域的其它显著表达特征纳入后,看目标HERV是否还能解释GWAS信号
- 一个GWAS locus里可能有多个gene和HERV都显著,因为它们的预测表达模型共享同一批LD相关SNP。条件分析会把同一区域的显著表达特征放在一起,估计谁还能在联合模型中保留信号
- 这个HERV显著,是因为它自己能解释GWAS信号,还是因为它和旁边某个gene的预测表达高度相关?
- 如果还能解释,它就更像是独立信号;如果不能,可能只是“蹭到”附近gene或LD结构
- 方法:还是用FUSION,它不是拿真实表达矩阵直接做条件分析,而是基于遗传预测表达
- 比如
HERV1预测表达 = 0.3×SNP1 + 0.1×SNP2 - 0.2×SNP3 GeneA预测表达 = 0.2×SNP1 + 0.4×SNP2 + 0.1×SNP4- FUSION用SNP weights和LD矩阵计算二者预测表达的相关性
corr(HERV1预测表达, GeneA预测表达),判断HERV1和GeneA是不是由同一批或高度相关的一批SNP预测出来的,如果相关性很高,那么它们的TWAS信号可能来自同一个遗传信号;如果相关性低,则更可能是独立信号 - 之后FUSION条件/联合分析会进一步做类似这样的模型
disease GWAS signal ~ predicted GeneA expression + predicted HERV1 expression + predicted GeneB expression,然后看每个feature在联合模型中的P值,也就是论文中说的joint P(普通rTWAS是每个feature单独看HERV1 ~ disease risk/GeneA ~ disease risk,也称marginal association边际关联) - 如果HERV1单独显著,联合模型里也显著,作者就认为它是一个conditionally independent association
- 比如
- 用FOCUS在每个LD block内做TWAS fine-mapping(精细定位):在一个LD block中比较多个候选表达特征,估计谁最可能是因果信号。条件分析回答“是否独立”,回答“在同一区域多个候选表达特征中,谁更可能是因果/主解释信号”
- PIP越接近1,说明该表达特征越可能是该区域的主解释信号
- 条件分析后的局部区域图:每个小图上半部分是基因组上下文,下半部分是GWAS变异关联信号。灰点是原始GWAS中每个SNP与疾病风险的边际关联P值,蓝点是在条件化该locus中联合显著表达特征之后 每个SNP剩余的GWAS关联P值
- 如果蓝点明显比灰点低,说明:原始SNP→疾病风险信号有一部分和gene/HERV预测表达信号重合
- GWAS峰:Manhattan plot上纵轴是
-log10(p),p值越小点越高。如果在某个基因组区域里,一群相邻SNP都和疾病显著相关,在图上形成一个像山峰一样的区域- 为什么一个区域会有“一群SNP”同时显著:连锁不平衡(linkage disequilibrium, LD)现象——附近SNP常常一起遗传。真正影响疾病风险的可能只有其中一个变异,但它周围一串SNP因为和它一起遗传,也会表现出疾病关联
- 所以GWAS峰通常说明:这个区域有遗传风险信号,但不一定立刻知道到底是哪个SNP、哪个gene或哪个HERV在起作用
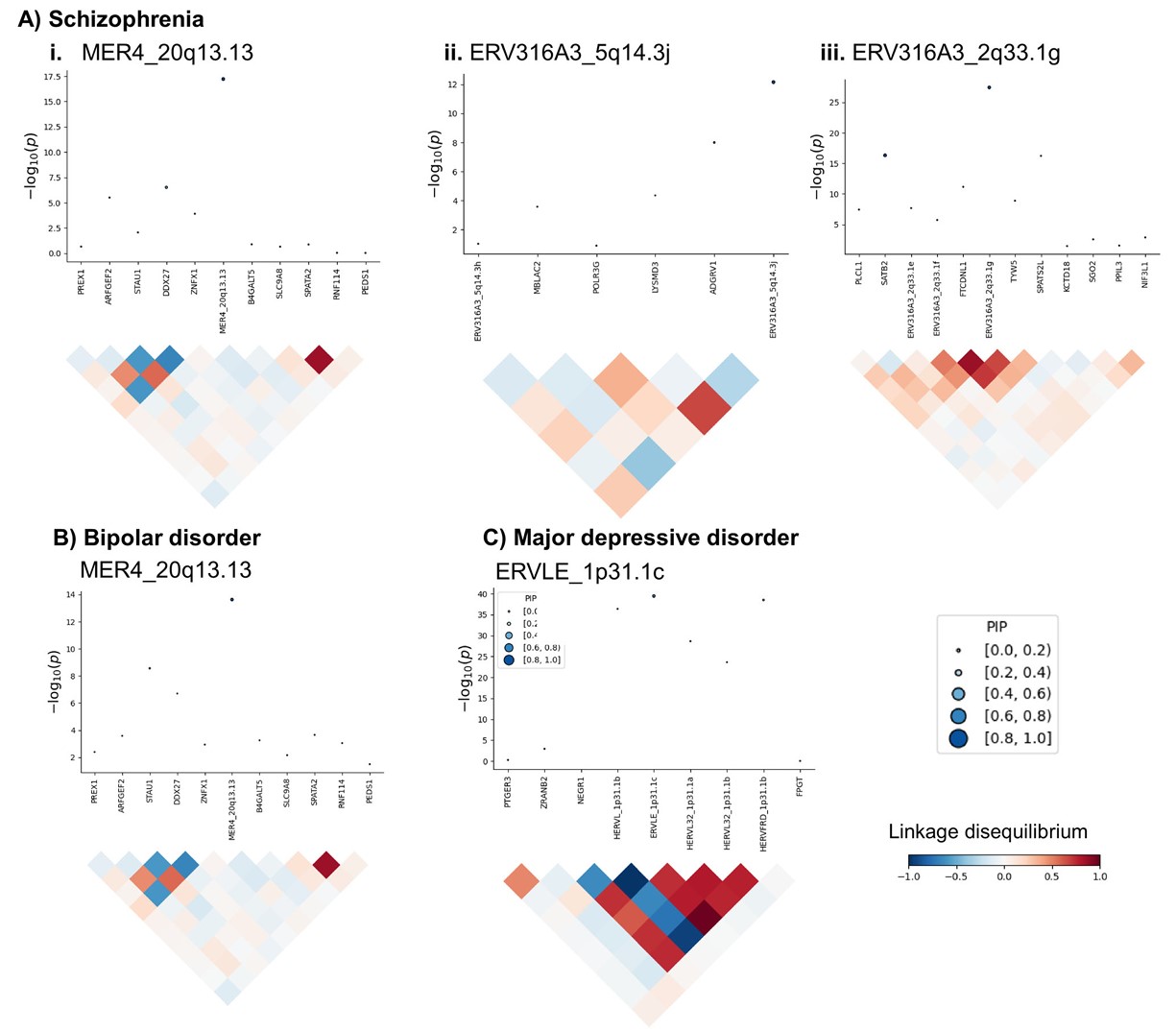
- 上半部分每个点是一个表达特征,纵轴是TWAS关联p值;点的大小和颜色代表PIP;下半部分是这些用遗传变异预测出来的表达特征的相关性
- 预测表达特征:比如说一个hERV位点和基因的SNP weights模型分别为
HERV1预测表达 = 0.3×SNP1 + 0.1×SNP2 - 0.2×SNP3和GeneA预测表达 = 0.2×SNP1 + 0.4×SNP2 + 0.1×SNP4,预测表达是根据一个人的基因型推算出来的表达倾向,预测表达特征就是这些SNP位点,表达特征的相关性就是“hERV1和geneA的遗传预测表达是不是由相似的一批SNP决定”,如果它们相关性很高,说明这两个表达特征的TWAS信号可能来自相同或相近的遗传信号 - 当然预测表达特征和LD有关:即使MER4和GeneA没有使用完全相同的SNP,但它们的SNP高度LD,那么它们的预测表达也会相关
- 因此,
预测表达相关性=SNP weights+SNP之间LD结构共同决定的相关性 - 如果两个feature间是正相关,就说明“预测HERV表达升高的遗传背景也倾向于预测GeneA表达升高”。以ERVLE_1p31.1c区域为例,它出现大片红/蓝结构,说明这个区域里很多候选表达特征的预测表达彼此相关或反相关,这意味着这个区域里多个gene/HERV的rTWAS信号可能互相牵连,不能只看谁的rTWAS P值最小;或者如果两个表达特征的预测表达高度相关,TWAS里它们可能都会显著,这时很难说到底是GeneA还是HERV1在解释GWAS信号,这些时候就要用FOCUS fine-mapping来判断谁更可能解释局部遗传信号
- 预测表达特征:比如说一个hERV位点和基因的SNP weights模型分别为
- 作者把“条件分析显著”且“PIP>0.5”的HERV定义为high confidence risk HERVs
GWAS → TWAS/rTWAS → 条件分析 → fine-mapping:一个逐层筛选、逐层提高可信度的流程
- GWAS:
SNP → 疾病风险,比如某个区域里很多SNP都和精神分裂症显著相关,于是形成一个GWAS峰,我知道“这个基因组区域和疾病风险有关”,但不知道它是通过哪个功能元件起作用的,可能是geneA/geneB/hERV/增强子,或者只是LD牵连- “GWAS发现的风险变异多数是非编码的,因此通常被认为可能影响局部基因调控”
- TWAS/rTWAS:
SNP → HERV/gene预测表达+SNP → 疾病风险,如果同一批SNP既能预测某个HERV/gene表达,又和疾病风险有关,TWAS就会说“这个HERV/gene的遗传预测表达与疾病风险相关”- 不是说“病人真实表达升高/降低”,而是“遗传上预测的表达高低与疾病风险相关”
- 条件分析:与疾病风险相关的HERV/gene是它独立影响的,还是借用了邻近gene的影响力?或者说,HERV1在考虑GeneA/GeneB/GeneC之后,是否还显著?如果还显著,就是我们想要的真正有影响力的hERV位点
- fine-mapping:在这个LD区域里,它是不是最可能解释该TWAS/GWAS信号的表达特征之一
- high confidence risk HERVs:
rTWAS显著+条件分析中独立+fine-mapping中PIP>0.5
第5步:敏感性分析和祖源外推
- 只用欧洲未患病对照构建TWAS weights,再与完整欧洲样本构建的weights比较
- 加入病例后,样本量增加133%
- 检测到cis-heritable HERV数量增加85%
- 用对照only权重和全样本权重做精神分裂症rTWAS,Z-score高度相关,Pearson r=0.95,但对照only分析显著结果减少16%
- 结论;结果不是明显由病例状态造成的偏差,把病例纳入建模不是为了做病例-对照差异,而是为了增加样本量、提高cis表达调控建模能力
- 尝试跨祖源验证,但结果较弱
- MER4_20q13.13在亚洲精神分裂症GWAS中有名义显著关联,但不能通过多重检验
- 用非洲血统CMC权重和非洲裔精神分裂症GWAS时,没有Bonferroni显著表达信号,作者认为主要是GWAS功效不足
- 结论;目前高可信结果主要限于欧洲血统,不能直接推广到其它祖源
第6步:这些高可信HERV在基因组上是什么关系
- 这些HERV信号到底是独立HERV转录,还是普通gene转录本的一部分
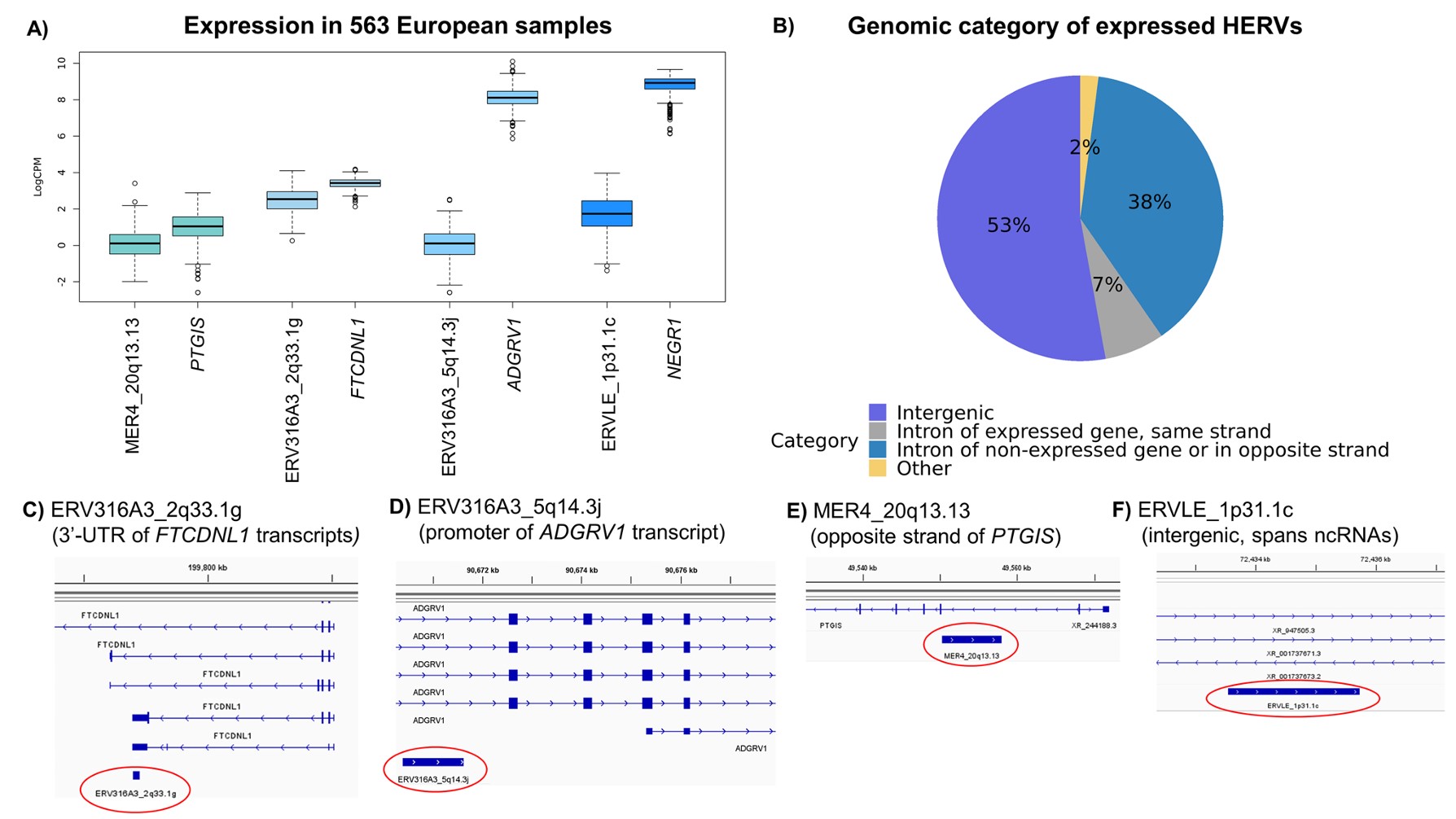
- A:比较高可信HERV和最近常规基因的logCPM表达,结果显示HERV表达普遍低于附近gene。作者认为这符合成人脑中HERV通常受表观遗传抑制的认识
- B:用HOMER注释HERV所在基因组区域,发现约98%的表达HERV位于基因间区或内含子区域
- intronic HERV:位于某个gene内含子中,不一定就是该gene转录本的一部分
- 如果HERV和gene同链且gene表达强,可能是gene isoform(基因mRNA前体剪接产生多种mRNA)的一部分;如果反义链、gene不表达或位于基因间区,更可能是独立非编码RNA
- C-D:ERV316A3_2q33.1g与FTCDNL1转录本3’UTR重叠;ERV316A3_5q14.3j位于ADGRV1转录本启动子区域。作者据此认为这两个rTWAS信号可能反映FTCDNL1和ADGRV1的特定isoform
- 这类HERV信号不能简单解释为“独立HERV RNA”,也可能是HERV参与形成的gene转录本结构。
- E-F:MER4_20q13.13位于PTGIS反义链;ERVLE_1p31.1c位于基因间区,最近基因是NEGR1,附近有ncRNA注释。作者认为这两个更可能代表新的非编码RNA
第7步:HERV可能参与哪些功能网络
- 在欧洲血统563个DLPFC样本中,把常规基因和HERV一起做WGCNA,得到16个表达模块,另有grey模块表示无法归类的特征
- 所有模块都包含HERV,说明HERV表达并不只集中在某个单一网络中,而是分布在多个表达程序里
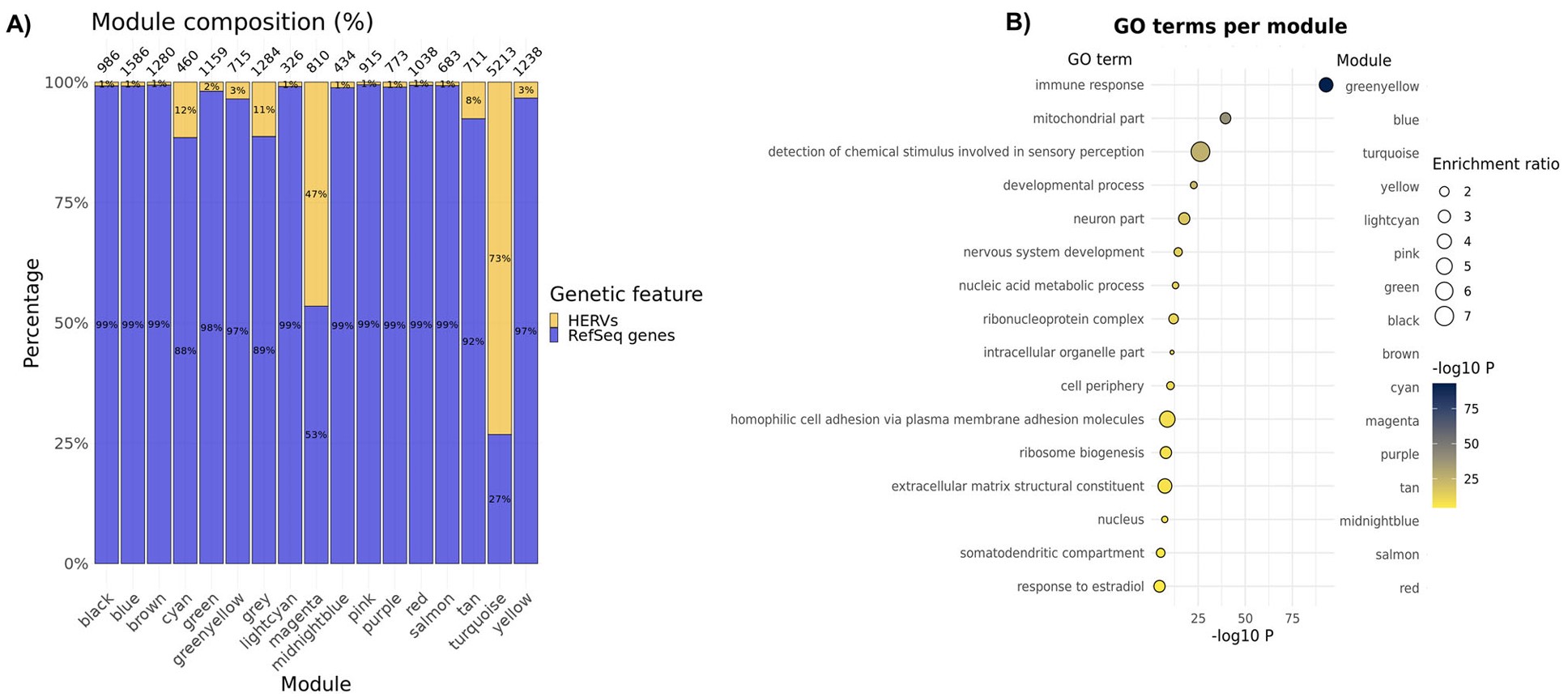
- A:每个共表达模块中gene和HERV的比例
- B:每个模块的GO富集结果
- cyan模块:synapse相关
- blue模块:mitochondria相关
- greenyellow模块:immune response相关
- turquoise模块:signal transduction相关
- 作者特别强调turquoise模块:包含1398个常规基因,占27%;包含3815个HERV,占73%;包括Table2中的所有高可信risk HERV;富集于信号转导相关GO term,如G protein-coupled receptor activity和detection of chemical stimulus
- 还做了两类稳健性检查:
- 调整样本来源、RIN、性别、病例状态、PMI、年龄、祖源PC和surrogate variables后,结果类似
- 在非洲血统229个样本中平行分析,turquoise模块也主要包含HERV,并且同样支持信号转导相关功能
- 结论:高可信HERV并不是孤立表达,而是落在一个HERV高度富集、并与信号转导相关gene共表达的大模块中
研究方法总结:遗传风险变异是否通过影响DLPFC中某些HERV位点的表达,从而参与精神疾病风险?
- RNA-seq定量gene和HERV表达
- 基因型数据建立表达预测模型(SNP weights),筛出能被cis遗传变异预测的HERV
- 把这些预测模型和精神疾病GWAS整合,做rTWAS,看看哪些HERV/gene的遗传预测表达与疾病风险相关(SNP~HERV/gene预测表达 + SNP~疾病风险)
- 用条件分析排除邻近gene牵连,看看哪些hERV位点在考虑邻近基因后还有影响力
- 用fine-mapping进一步判断哪个候选HERV是否最可能解释局部信号
- 用基因组上下文判断HERV是gene isoform的一部分还是独立ncRNA
- 用WGCNA从HERV和常规基因的共表达模块推断功能背景(GO富集)
为什么要分别分析欧洲和非洲:
- 欧洲和非洲血统是CMC中最大的、相对同质的两个子集:“同质”主要是遗传祖源上的同质,因为rTWAS需要用附近SNP预测表达,如果样本中混合很多祖源,SNP频率和LD结构会变复杂,预测模型会不稳定
- TWAS/rTWAS需要祖源匹配的SNP weights、LD reference和GWAS结果
- LD结构不同:某些SNP在欧洲人群中高度连锁,在非洲人群中可能不连锁
- 如果用欧洲weights去解释非洲GWAS,统计结果可能偏差
- 欧洲GWAS统计功效最高(欧洲精神疾病GWAS样本量更大、结果更稳定),所以用欧洲分析作为主分析
- 非洲样本用于两个目的:看HERV调控是否有祖源差异,以及测试结果能否外推
- 在表达结果中,非洲血统样本表达HERV数量与欧洲样本接近,但cis遗传调控的HERV重叠并不完全(非洲样本中852个cis-heritable HERV里,只有534个也在欧洲样本中显示cis调控)。虽然需要谨慎解释,因为两组样本量不同,但这提示可能存在祖源相关的HERV表达调控差异
- 测试欧洲样本中发现的高可信HERV是否能推广到其它祖源:跨祖源使用欧洲weights并不理想,因为不同人群LD结构不同
主要结论:
- 在DLPFC中检测到大量HERV表达,并且一部分受cis遗传调控
- 在欧洲血统563个样本中,作者检测到4594个表达HERV,其中4289个位于常染色体,1238个有cis-heritable expression
- 在非洲血统229个样本中,检测到4645个表达HERV,其中852个有cis-heritable expression
- 两组中表达的canonical genes数量接近,但欧洲组中检测到更多cis调控表达特征,可能与样本量更大有关
- rTWAS发现多个精神疾病相关的HERV表达信号
- 精神分裂症中发现163个Bonferroni显著表达特征,其中15个是HERV
- 双相情感障碍中发现47个显著表达特征,其中2个是HERV
- 重度抑郁障碍中发现29个显著表达特征,其中9个是HERV
- ADHD和自闭症谱系障碍中没有发现HERV显著信号
- 条件分析筛出独立于邻近gene的HERV信号
- 精神分裂症中有91个条件独立表达关联,其中6个是HERV
- 双相障碍中有30个条件独立关联,其中2个是HERV
- 重度抑郁障碍中有12个条件独立关联,其中2个是HERV
- fine-mapping进一步筛出高可信risk HERV
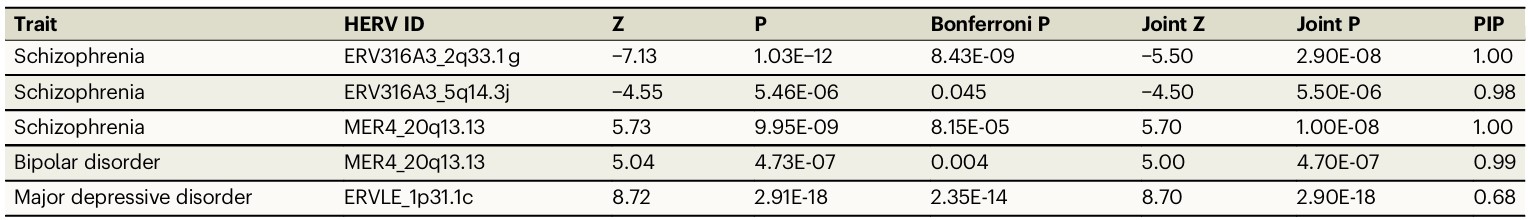
- 精神分裂症中有3个高可信HERV:ERV316A3_2q33.1g、ERV316A3_5q14.3j、MER4_20q13.13
- 双相障碍中MER4_20q13.13也是高可信HERV
- 重度抑郁障碍中ERVLE_1p31.1c是高可信HERV
- 把病例纳入权重构建并没有明显改变结果方向,主要作用是提高检测cis调控和rTWAS信号的统计功效:
- 加入病例使样本量增加133%,cis-heritable HERV检测增加85%
- 只用欧洲未患病对照做精神分裂症rTWAS时,结果与全样本weights高度相关,但显著信号减少16%
- 高可信HERV可能有不同分子含义
- HERV表达普遍低于附近gene
- 大多数表达HERV位于基因间区或内含子区
- ERV316A3_2q33.1g与FTCDNL1转录本3’UTR重叠,ERV316A3_5q14.3j位于ADGRV1转录本启动子区域,因此它们可能反映特定gene isoform
- 而MER4_20q13.13位于PTGIS反义链,ERVLE_1p31.1c位于基因间区,作者推测它们更可能是ncRNA
- 部分高可信HERV与预测增强子区域重叠,但需要实验验证
- 在UCSC Browser中查看发现ERVLE_1p31.1c、ERV316A3_2q33.1g和MER4_20q13.13所在位置包含ENCODE预测远端增强子,但ERV316A3_5q14.3j附近没有
- 由于缺少长读长RNA-seq来精确定义HERV转录本位置,增强子预测也需要实验验证
- 高可信risk HERV并不是孤立表达信号,而是落在一个HERV高度富集、与信号转导相关的共表达模块中
2024 NC–schERV+eQTL
- 计数
- scRNA-seq:包含981个供体和约120万个细胞
- 去掉了与基因外显子重叠的HERV位点,cellranger 构建hERV+gene注释直接计数hERV,也是位点级
- 只保留unique mapping reads
- 标准化:(HERV计数×1e4)/(总计数),保留至少了20个细胞中表达的hERV
- 用了Pseudobulk
- 用常规基因表达注释PBMC细胞类型,识别细胞类型特异HERV,最后识别出1936个细胞类型特异HERV
- 用表观遗传数据解释细胞类型特异性:把细胞类型特异HERV与Roadmap/ENCODE的chromatin state、H3K27ac、H3K4me3等数据整合,发现这些HERV富集在对应细胞类型的active chromatin、enhancer或promoter区域。把“这个HERV在某类细胞中特异表达”进一步解释为“这个HERV所在区域在该细胞类型中处于开放/活跃调控状态”
- Pseudobulk计数矩阵+TensorQTL做cis-eQTL分析;用SMR分析三类关系:variant-eHERV-eGene、variant-eHERV-disease、variant-eGene-disease;再用coloc做GWAS和eQTL共定位验证。某个遗传变异影响HERV表达,HERV表达与附近疾病相关基因或疾病GWAS信号有关
后续思路
都是正常组织 建SNP和hERV的关系 从GWAS/eQTL(孟德尔随机化)推疾病和hERV的关系
- 要拿外部数据库去推断 没有从实际数据上去看
- 重点——SNP~hERV~疾病:
- 单细胞中大脑皮层的SNP和hERV关系
- epi连接SNP和hERV(SNP在调控元件上,影响不同hERV,联系到疾病中hERV的变化)
- hERV与疾病的因果关系(找mediation):hERV调控转录本导致疾病
- SNP-hERV-疾病:直接overlap/SMR算显著hERV
- 疾病里面失调的TF(SNP影响的TF)同时影响hERV和gene:LTR上结合的TF是哪些
- lncRNA领域:hERV通过miRNA(SNP影响的miRNA)影响基因
表观遗传:解释hERV为什么发生变化,作为结果的补充 组蛋白乙酰化/染色质可及性影响比较大,甲基化比较小
计划安排
- 沿着单细胞mRNA这条路,仿照那篇NC,把功能、疾病那块做清楚:扩展多个数据集,下游分析有套路,结果意义中等
- 考虑hERV尤其是LTR作为enhancer、调控的角度去做与AD疾病的关系:新意、意义上更大一点
- 一起整合起来做也行
Single-cell eQTL mapping:HERV不是简单的基因组残留物,它们在不同免疫细胞中是否有特异表达?这些表达是否受遗传变异调控?是否和疾病风险有关?
- 在单细胞RNA-seq中重新计数HERV位点表达:使用特殊的筛选方法,尽量只保留“独立转录的HERV”(hERV计数方法?)
- 判断HERV表达是否真的有细胞类型特异性:HERV表达谱在不同免疫细胞之间差异明显,而且HERV表达与邻近基因整体相关性较低,说明很多HERV信号不是简单来自附近基因转录本的“顺带计数”(如何区分“某个hERV位点到底是自己转录,还是只是落在某个基因转录本里”)
- HERV-eQTL分析:把供体基因型和各细胞类型中的HERV表达联系起来,看哪些SNP会影响某个HERV位点的表达(eQTL分析需要同一批样本的基因型数据,现在的数据集没有这方面数据)
- HERV-eQTL和疾病GWAS整合:用免疫相关疾病GWAS做SMR分析,检验“变异-HERV-疾病”、“变异-HERV-基因”、“变异-基因-疾病”这几类关系,从而提出某些HERV可能介导遗传变异与自身免疫疾病风险之间的联系
snATAC分析:这个细胞的哪些基因组区域是开放的、可能正在参与调控
- 给细胞类型做表观遗传注释:GSE174367原文在snATAC中识别了兴奋性神经元、抑制性神经元、星形胶质细胞、小胶质细胞、少突胶质细胞和OPC等主要细胞类型
- 找AD相关开放区域:比较AD和对照,在某类细胞里哪些peak更开放或更关闭。GSE174367原文识别了细胞类型特异、疾病相关的候选顺式调控元件,并把这些元件和候选靶基因联系起来
- 找转录因子调控线索:看某些转录因子的结合位点是否在AD中更开放。GSE174367原文做了TF motif变异分析,并在不同细胞类型中寻找疾病相关TF调控信号
- 和GWAS风险位点整合:很多AD风险变异不在编码区,而在调控区,snATAC可以帮助判断这些风险位点在哪类细胞中开放,可能调控哪些基因。GSE174367原文把AD GWAS精细定位信号与snATAC数据整合,用来连接风险信号、细胞类型和顺式调控关系
- 在我的课题里:
- AD中差异表达的hERV/LTR位点,附近染色质是否也更开放
- 某些LTR是否本身就是AD相关开放peak
- 这些开放的LTR是否可能作为enhancer/promoter影响附近AD相关基因
下周文献汇报Single-cell eQTL mapping以及相关文章,学习并实践一下snATAC分析,因为我现在手里有snATAC数据,可以先尝试一下。转录组这一块我之前挖的已经比较深了,只剩下一些小问题,可能还要再找新数据
其它
gtf文件gene行染色体范围和基因的重叠,比较是不是重叠多的表达高
我的表达多的类别是不是别人也报道了,和total RNA的报道的是不是相同;ERV位点在基因组内的分布,是不是分布多的表达也多
人体ncRNA什么情况下会发生ployA加尾 **